Scalable Synthesis of LND1067 Antibody-Drug Conjugate Linker for Commercial Production
The rapidly evolving landscape of oncology therapeutics has placed Antibody-Drug Conjugates (ADCs) at the forefront of targeted cancer therapy. Central to the efficacy of these complex biologics is the linker technology, which ensures the stable transport of cytotoxic payloads to tumor sites while minimizing systemic toxicity. Patent CN113336823A discloses a breakthrough synthetic methodology for LND1067, a critical linker-drug conjugate featuring the GGFG peptide sequence and the potent topoisomerase I inhibitor Dxd. This innovation addresses long-standing challenges in ADC manufacturing by providing a route that is not only chemically robust but also optimized for high yield and scalability. As the demand for reliable antibody-conjugated drug linker suppliers intensifies, this technology represents a pivotal advancement for pharmaceutical developers seeking to secure their supply chains for next-generation ADCs.
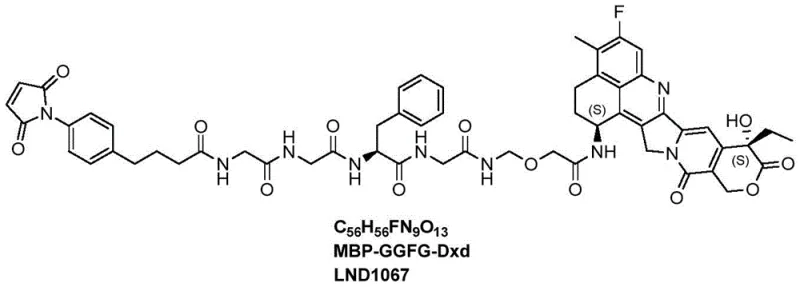
The structural complexity of LND1067, comprising a maleimide moiety for antibody conjugation, a protease-cleavable peptide linker, and the exatecan derivative payload, necessitates a synthesis strategy that meticulously controls stereochemistry and purity. The disclosed method achieves this through a convergent approach that minimizes step count and maximizes material throughput. By integrating advanced peptide coupling techniques with precise deprotection strategies, the process ensures the production of high-purity intermediates essential for clinical-grade ADCs. This technical leap forward underscores the importance of adopting novel synthetic routes to meet the rigorous quality standards of the biopharmaceutical industry.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of linkers similar to LND1067 has been plagued by inefficiencies associated with multi-step protection and deprotection sequences. Traditional routes often rely on the preparation of specific intermediates, such as LND1067-2-1, which serve as precursors for the final conjugation. As illustrated in the prior art, these conventional pathways frequently suffer from suboptimal reaction yields at critical junctions. The synthesis of the intermediate LND1067-2-1, in particular, has been identified as a bottleneck, exhibiting low conversion rates that directly compromise the overall efficiency of the process. Furthermore, the accumulation of impurities during these extended sequences complicates downstream purification, often requiring extensive chromatographic separation that drives up manufacturing costs and extends lead times.
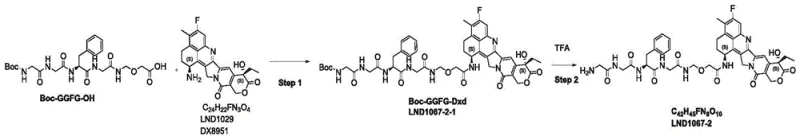
In addition to yield issues, conventional methods often employ reaction conditions that are difficult to control on a large scale. The use of sensitive protecting groups that require harsh removal conditions can lead to degradation of the potent cytotoxic payload, resulting in reduced potency and increased safety risks. The reliance on intermediates with poor stability further exacerbates supply chain vulnerabilities, as storage and transportation become critical failure points. For procurement managers and supply chain heads, these limitations translate into unpredictable availability and inflated costs for high-purity ADC intermediates, hindering the rapid development and commercialization of life-saving therapies.
The Novel Approach
The synthetic method described in patent CN113336823A introduces a streamlined, high-efficiency pathway that circumvents the pitfalls of traditional chemistry. By utilizing LND1067-L1 (an Fmoc-protected peptide acid) and LND1029 (the Dxd derivative) as key starting materials, the new route eliminates the need for the problematic LND1067-2-1 intermediate entirely. This strategic redesign allows for a direct coupling reaction mediated by HATU, which proceeds with exceptional efficiency under mild conditions. The subsequent removal of the Fmoc protecting group using diethylamine is rapid and clean, avoiding the side reactions often associated with acidic or basic deprotection methods. This novel approach not only enhances the chemical yield but also significantly simplifies the operational workflow.
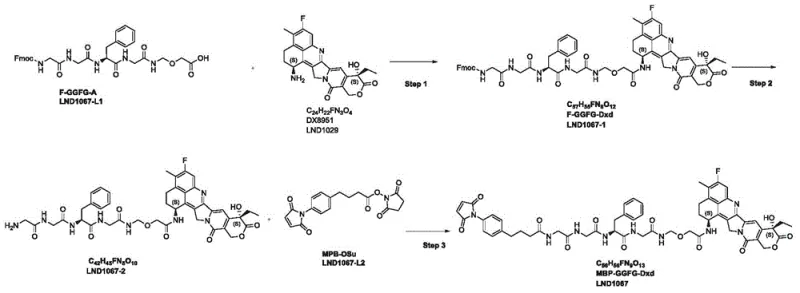
Furthermore, the final conjugation step with LND1067-L2 (MPB-OSu) is optimized to ensure high conversion while maintaining the integrity of the maleimide ring, which is crucial for subsequent antibody attachment. The entire process is supported by a robust purification protocol using medium-pressure reverse-phase chromatography, which effectively removes trace impurities and reagents. This level of process control is vital for meeting the stringent regulatory requirements for ADC components. For R&D directors, this novel approach offers a viable path to scalable manufacturing, reducing the technical risk associated with process transfer from laboratory to commercial production facilities.
Mechanistic Insights into HATU-Mediated Peptide Coupling and Fmoc Deprotection
The core of this synthetic success lies in the mechanistic precision of the coupling and deprotection steps. The initial formation of LND1067-1 relies on the activation of the carboxylic acid group of LND1067-L1 by HATU (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate). This uronium-based reagent generates a highly reactive O-acylisourea intermediate, which rapidly reacts with the amine group of LND1029 to form the stable amide bond. The presence of DIPEA as a base facilitates this process by neutralizing the generated acid and maintaining the nucleophilicity of the amine. Conducting this reaction at low temperatures (0-5°C) is critical to suppress racemization and minimize side reactions, ensuring the stereochemical purity of the chiral centers within the peptide backbone.
Following the coupling, the removal of the Fmoc group from LND1067-1 to generate LND1067-2 proceeds via a beta-elimination mechanism triggered by the secondary amine, diethylamine. This reaction is highly specific and occurs under mild conditions that do not affect the sensitive ester or lactone moieties present in the Dxd payload. The use of diethylamine, rather than stronger bases like piperidine, offers a balance between reaction rate and selectivity, preventing potential degradation of the complex molecular architecture. This mechanistic understanding allows for fine-tuning of reaction parameters, such as stoichiometry and temperature, to maximize yield and minimize the formation of deletion sequences or byproducts, thereby ensuring a clean impurity profile for the final API intermediate.
How to Synthesize LND1067 Efficiently
The synthesis of LND1067 is executed through a logical three-step sequence that prioritizes yield and purity at every stage. The process begins with the coupling of the peptide linker to the toxin payload, followed by deprotection to reveal the reactive amine, and concludes with the attachment of the antibody-binding maleimide handle. Each step is monitored by HPLC to ensure completion before proceeding, and purification is achieved via reverse-phase chromatography to guarantee pharmaceutical-grade quality. The detailed standardized synthetic steps for this high-efficiency route are outlined below.
- React LND1067-L1 (Fmoc-GGFG-A) with LND1029 (DX8951) using HATU and DIPEA in DMF at 0-5°C to form LND1067-1.
- Deprotect LND1067-1 by reacting with Diethylamine (DEA) in DMF at 0-5°C to remove the Fmoc group, yielding LND1067-2.
- Conjugate LND1067-2 with LND1067-L2 (MPB-OSu) using DIPEA in solvent at -5 to 0°C, followed by reverse-phase purification to obtain LND1067.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this novel synthetic route offers tangible benefits that extend beyond mere chemical elegance. The elimination of the low-yield intermediate LND1067-2-1 fundamentally alters the cost structure of LND1067 production. By removing a bottleneck step that previously suffered from poor conversion, the overall material throughput is significantly increased, leading to substantial cost savings in raw material consumption. Furthermore, the simplified workflow reduces the number of unit operations required, lowering labor and utility costs associated with manufacturing. This efficiency gain translates directly into a more competitive pricing model for high-purity ADC linkers, enabling pharmaceutical companies to optimize their R&D budgets.
- Cost Reduction in Manufacturing: The new method achieves markedly higher yields in the initial coupling step (reported up to 90% in examples versus 60% in comparative methods), which drastically reduces the cost of goods sold (COGS). By avoiding the synthesis of unstable intermediates and utilizing robust reagents like HATU and DIPEA, the process minimizes waste generation and the need for expensive rework or recycling of materials. This lean manufacturing approach ensures that cost reduction in ADC manufacturing is realized through fundamental process improvements rather than superficial adjustments.
- Enhanced Supply Chain Reliability: The robustness of the synthetic route enhances supply chain continuity by reducing the risk of batch failures. The use of stable starting materials and mild reaction conditions ensures consistent quality across different production batches, mitigating the risk of supply disruptions caused by out-of-specification products. Additionally, the streamlined process shortens the overall production cycle time, allowing for faster response to market demands and reducing lead time for high-purity pharmaceutical intermediates. This reliability is crucial for maintaining the tight schedules of clinical trials and commercial launches.
- Scalability and Environmental Compliance: The method is explicitly designed for commercial scale-up, utilizing solvents and reagents that are compatible with large-scale reactor systems. The purification strategy, based on reverse-phase chromatography, is readily scalable and avoids the use of hazardous heavy metal catalysts often found in alternative coupling methods. This aligns with modern environmental, health, and safety (EHS) standards, facilitating easier regulatory approval and reducing the environmental footprint of the manufacturing process. The ability to scale from grams to kilograms without significant process re-engineering makes this an ideal solution for growing ADC programs.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and supply of LND1067. These answers are derived directly from the technical specifications and experimental data provided in the patent literature, ensuring accuracy and relevance for industry professionals. Understanding these details is essential for evaluating the feasibility of integrating this linker into your ADC development pipeline.
Q: What is the primary advantage of the new LND1067 synthesis method over conventional routes?
A: The novel method bypasses the synthesis of the low-yield intermediate LND1067-2-1 required in traditional routes. By utilizing an Fmoc-protected starting material (LND1067-L1) and direct coupling with the toxin payload, the process achieves significantly higher overall yields (up to 90% in initial steps compared to 60% in comparative examples) and simplifies the purification workflow.
Q: What purification technique is recommended for LND1067 intermediates?
A: The patent specifies the use of medium-pressure reverse-phase chromatography. For LND1067-1 and LND1067-2, a 0.1% TFA water/acetonitrile system is used. For the final LND1067 product, a 0.1% AcOH water/acetonitrile system is employed, ensuring high purity suitable for pharmaceutical applications.
Q: Is this synthesis method suitable for large-scale manufacturing?
A: Yes, the method is explicitly designed for scale-up. It utilizes robust coupling reagents like HATU and standard solvents (DMF, DCM), avoids unstable intermediates, and employs straightforward workup procedures such as lyophilization and reverse-phase purification, making it highly adaptable for commercial-scale production of ADC linkers.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable LND1067 Supplier
The technological advancements presented in patent CN113336823A highlight the critical role of innovative chemistry in the ADC sector. NINGBO INNO PHARMCHEM stands ready to leverage this expertise to support your drug development goals. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications required for clinical and commercial ADC intermediates. We understand that the quality of the linker dictates the safety and efficacy of the final conjugate, and we are committed to delivering excellence at every step.
We invite you to engage with our technical team to discuss how our optimized synthesis of LND1067 can accelerate your project timelines. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you navigate the complexities of ADC manufacturing with confidence and precision.