Advanced Synthesis of 1-Benzyl-2,4-diarylimidazoles for Next-Generation Antitumor Agents
The pharmaceutical industry is constantly seeking novel scaffolds to overcome drug resistance in oncology, particularly for breast cancer treatment. Patent CN114315729A introduces a robust chemical preparation method for 1-benzyl-2,4-diarylimidazole compounds, which function as potent HSP90 inhibitors. Unlike earlier iterations of HSP90 inhibitors that suffered from poor solubility or low oral bioavailability, these new structures demonstrate antitumor activity equivalent to or superior to the clinical benchmark 17-AAG. For R&D directors and procurement specialists, this patent represents a significant opportunity to access high-value pharmaceutical intermediates with a clear path to clinical relevance. The synthesis strategy outlined in this document provides a scalable and chemically elegant solution to a long-standing problem in heterocyclic chemistry, ensuring a reliable supply chain for next-generation anticancer agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 1-benzyl-2,4-diarylimidazoles has relied heavily on condensation reactions between acetophenone and benzylamine derivatives, often catalyzed by iodine, copper salts, or electrochemical methods. While these approaches are efficient for generating symmetric or simple structures, they suffer from a critical flaw: the inability to easily differentiate the substituents at the 2 and 4 positions of the imidazole ring. Furthermore, some legacy methods, such as those disclosed by Wang et al. in 1994, utilized toxic organotin reagents like trimethylphenyltin, posing severe environmental and safety hazards for large-scale API manufacturing. These limitations restrict the chemical diversity available to medicinal chemists and complicate the purification process due to the formation of regioisomers, ultimately driving up costs and extending lead times for drug development programs.
The Novel Approach
The methodology described in CN114315729A circumvents these issues through a modular, stepwise construction of the imidazole core. By starting with 4-bromoimidazole and employing sequential palladium-catalyzed cross-coupling reactions, the process allows for the independent introduction of distinct aryl groups at the C2 and C4 positions. This strategic disconnection ensures high regioselectivity and structural precision, which is essential for optimizing structure-activity relationships (SAR). The elimination of toxic tin reagents in favor of benign boronic acids not only aligns with green chemistry principles but also simplifies the downstream purification workflow. This novel approach transforms the production of these complex heterocycles from a challenging artistic endeavor into a predictable, industrial-grade chemical process suitable for commercial scale-up.
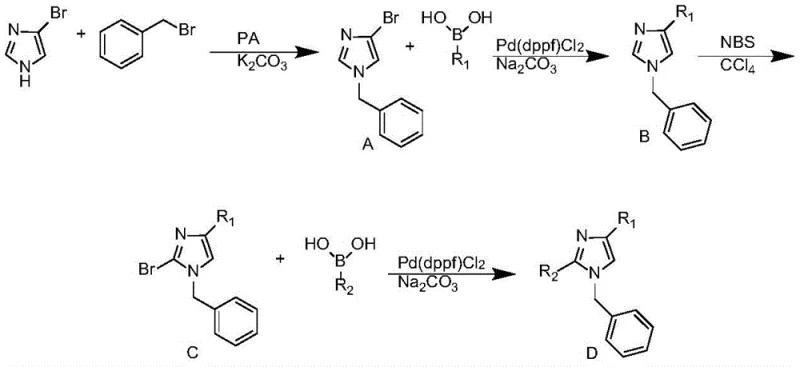
Mechanistic Insights into Palladium-Catalyzed Sequential Coupling
The core of this synthetic innovation lies in the precise manipulation of the imidazole ring's reactivity through palladium catalysis. The process initiates with the N-alkylation of 4-bromoimidazole using benzyl bromide and anhydrous potassium carbonate in acetone, establishing the N1-benzyl motif essential for biological activity. Subsequently, the first Suzuki-Miyaura coupling targets the C4 position. Utilizing Pd(dppf)Cl2 as the catalyst and sodium carbonate as the base in a 1,4-dioxane and water system, the reaction selectively couples the first arylboronic acid. The choice of Pd(dppf)Cl2 is critical here, as the dppf ligand stabilizes the palladium center, facilitating the oxidative addition and reductive elimination steps required for efficient C-C bond formation on the electron-deficient imidazole ring.
Following the installation of the first aryl group, the synthesis employs a regioselective bromination step using N-bromosuccinimide (NBS) in carbon tetrachloride. This step is mechanistically vital as it activates the C2 position for the second coupling event without affecting the newly formed C4-aryl bond. The final step involves a second Suzuki coupling with a different arylboronic acid under similar palladium-catalyzed conditions. This sequential strategy effectively decouples the substitution patterns at positions 2 and 4, allowing for the creation of diverse libraries of high-purity pharmaceutical intermediates. The rigorous control over reaction conditions, such as maintaining temperatures between 100-110°C during coupling and utilizing inert atmospheres, minimizes side reactions and ensures the integrity of the sensitive imidazole scaffold throughout the synthesis.
How to Synthesize 1-Benzyl-2,4-diarylimidazole Efficiently
The successful execution of this four-step sequence requires careful attention to stoichiometry and purification techniques to maximize yield and purity. The protocol begins with the alkylation of the imidazole nitrogen, followed by two distinct cross-coupling events separated by a halogenation step. Each stage builds upon the previous one, requiring precise control of molar ratios, such as the 1:2 to 1:3 ratio of substrate to boronic acid, to drive the reactions to completion. The detailed operational parameters, including solvent choices like 1,4-dioxane and specific workup procedures involving silica gel column chromatography, are designed to remove palladium residues and unreacted starting materials effectively. For a comprehensive guide on the exact experimental conditions and troubleshooting tips for each transformation, please refer to the standardized synthesis instructions below.
- Perform N-alkylation of 4-bromoimidazole with benzyl bromide using potassium carbonate in acetone to form Intermediate A.
- Execute the first Suzuki coupling at the C4 position using arylboronic acid and Pd(dppf)Cl2 catalyst to yield Intermediate B.
- Conduct regioselective bromination at the C2 position using NBS in carbon tetrachloride to generate Intermediate C.
- Complete the synthesis with a second Suzuki coupling at the C2 position using a different arylboronic acid to obtain the final Diarylimidazole product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this synthetic route offers tangible benefits beyond mere chemical elegance. The shift away from toxic organotin reagents to widely available arylboronic acids significantly reduces the regulatory burden and waste disposal costs associated with production. This change simplifies the sourcing of raw materials, as boronic acids are commodity chemicals with stable global supply chains, thereby enhancing supply chain reliability. Furthermore, the modular nature of the synthesis means that a single intermediate can be diverted to produce multiple final analogs by simply changing the boronic acid in the final step. This flexibility allows manufacturers to respond rapidly to changing market demands or clinical trial requirements without retooling the entire production line, offering substantial agility in a competitive landscape.
- Cost Reduction in Manufacturing: The elimination of expensive and hazardous tin reagents directly lowers the cost of goods sold (COGS). Additionally, the use of standard palladium catalysts and common solvents like acetone and ethyl acetate avoids the need for specialized equipment or exotic reagents. The streamlined purification process, which relies on standard silica gel chromatography rather than complex distillation or recrystallization from difficult solvents, further reduces operational expenses. By minimizing the number of unit operations required to achieve high purity, the overall manufacturing footprint is reduced, leading to significant cost savings in both material and labor resources.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as 4-bromoimidazole, benzyl bromide, and various substituted phenylboronic acids ensures that raw material availability is not a bottleneck. These precursors are produced by numerous suppliers globally, mitigating the risk of single-source dependency. The robustness of the Suzuki coupling reaction, which tolerates a wide range of functional groups, means that the process is less susceptible to batch-to-batch variability caused by minor fluctuations in raw material quality. This stability translates to more predictable production schedules and shorter lead times for delivering complex pharmaceutical intermediates to clients.
- Scalability and Environmental Compliance: The reaction conditions described, such as heating to 100-110°C in aqueous-organic biphasic systems, are readily transferable from laboratory glassware to industrial reactors. The absence of pyrophoric or highly toxic reagents simplifies safety protocols and reduces the need for specialized containment systems. Moreover, the aqueous workup steps facilitate the removal of inorganic salts and catalyst residues, ensuring that the final product meets stringent purity specifications required for GMP manufacturing. This alignment with environmental, health, and safety (EHS) standards makes the process highly attractive for sustainable fine chemical manufacturing and facilitates smoother regulatory approvals.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthesis is crucial for stakeholders evaluating its potential for integration into their supply chains. The following questions address common inquiries regarding the scalability, safety, and application of this technology. These answers are derived directly from the technical disclosures within the patent documentation, providing a factual basis for decision-making. By clarifying these points, we aim to demonstrate the viability of this route for producing high-quality antitumor agents that meet the rigorous demands of the modern pharmaceutical industry.
Q: What is the primary advantage of this synthesis method over traditional condensation routes?
A: Unlike traditional methods that condense acetophenone and benzylamine, this patented route utilizes sequential Suzuki couplings. This allows for the precise installation of different aryl substituents at the 2 and 4 positions of the imidazole ring, overcoming the regioselectivity limitations of prior art.
Q: How does this method address the toxicity concerns associated with older organotin protocols?
A: Previous methods, such as those reported by Wang et al. in 1994, relied on toxic trimethylphenyltin reagents. The current protocol replaces these hazardous materials with safer, commercially available arylboronic acids, significantly improving the environmental profile and safety of the manufacturing process.
Q: What represents the biological efficacy of these compounds compared to standard controls?
A: Biological evaluation demonstrates that specific derivatives, such as compounds D7, D8, and D11, exhibit antitumor proliferation inhibitory activities against MCF-7 breast cancer cells that are superior to the positive control 17-AAG, validating their potential as potent HSP90 inhibitors.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1-Benzyl-2,4-diarylimidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic route described in CN114315729A for developing novel HSP90 inhibitors. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from preclinical research to clinical supply. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 1-benzyl-2,4-diarylimidazole intermediate meets the highest quality standards. We are committed to supporting your drug discovery efforts with reliable, high-performance chemical solutions that accelerate your timeline to market.
We invite you to collaborate with our technical team to explore how this advanced synthesis can optimize your specific drug development program. By leveraging our expertise in palladium-catalyzed couplings and heterocyclic chemistry, we can provide a Customized Cost-Saving Analysis tailored to your volume requirements. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you secure a sustainable and cost-effective supply of these critical antitumor intermediates, ensuring your pipeline remains robust and competitive in the global oncology market.