Advanced Manufacturing of Fluorinated Benzopyran Intermediates via Streamlined Thermal Cyclization
Advanced Manufacturing of Fluorinated Benzopyran Intermediates via Streamlined Thermal Cyclization
The pharmaceutical industry constantly seeks more efficient pathways for complex heterocyclic scaffolds, particularly those containing fluorine substituents which are critical for metabolic stability and binding affinity. Patent CN1259941A discloses a groundbreaking process for the manufacture of 3-N,N-dicyclobutylamino-8-fluoro-3,4-dihydro-2H-1-benzopyran-5-carboxamide and its enantiomers. This technology represents a paradigm shift from previous methodologies by utilizing a starting material that already possesses the requisite fluorine substitution pattern. By circumventing the need for late-stage fluorination, this process not only enhances safety profiles by eliminating hazardous fluorinating agents but also drastically improves the overall atom economy. For R&D directors and procurement specialists, this patent offers a compelling alternative to the cumbersome routes described in prior art such as WO 95/11891, promising a more reliable supply of high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fluorinated benzopyran derivatives relied heavily on introducing the fluorine atom at a late stage of the synthesis, a strategy fraught with technical and economic challenges. The method disclosed in WO 95/11891, for instance, necessitates a selective bromination followed by a halogen-lithium exchange reaction, which must be conducted at cryogenic temperatures to maintain control over reactivity. This requirement for extreme cold imposes significant energy costs and limits the scalability of the process in standard reactor setups. Furthermore, the subsequent reaction with fluorinating agents often generates substantial quantities of non-fluorinated by-products, such as (R)-3-N,N-dibenzylamino-5-methoxy-3,4-dihydro-2H-1-benzopyran, which are structurally similar to the desired product. Separating these impurities typically requires expensive and technically difficult chromatographic methods, creating a bottleneck in production throughput and driving up the cost of goods significantly.
The Novel Approach
In stark contrast, the process outlined in CN1259941A adopts a "build-from-fluorine" strategy, commencing with 4-fluoro-3-hydroxybenzoic acid (Compound II), a commercially available feedstock. This strategic choice eliminates the entire sequence of bromination, lithiation, and fluorination, thereby removing the associated safety risks and equipment constraints. The new route leverages a thermal cyclization of a propargyl ether intermediate to construct the benzopyran core, a transformation that is robust and scalable. By shifting the complexity to the early stages of the synthesis where purification is easier and cheaper, the overall process efficiency is markedly improved. This approach not only reduces the total number of reaction steps but also ensures that the fluorine atom is retained throughout the synthesis without the risk of defluorination or scrambling, resulting in a superior impurity profile for the final active pharmaceutical ingredient intermediate.
Mechanistic Insights into Thermal Cyclization and Functionalization
The core of this innovative synthesis lies in the construction of the benzopyran ring system via a thermal rearrangement mechanism. The process begins with the esterification of the starting acid to form Compound (III), followed by O-alkylation with propargyl bromide to yield the propargyl ether (IV). Upon heating Compound (IV) to temperatures between 210°C and 230°C, a Claisen-type rearrangement occurs, followed by an intramolecular cyclization to form the chromene ester (V). This thermal step is particularly advantageous as it proceeds without the need for transition metal catalysts, which can often leave trace metal residues that are difficult to remove and toxic to biological systems. The absence of metal catalysts simplifies the downstream workup, requiring only standard aqueous washes and crystallization to achieve high purity levels, which is a critical factor for regulatory compliance in pharmaceutical manufacturing.
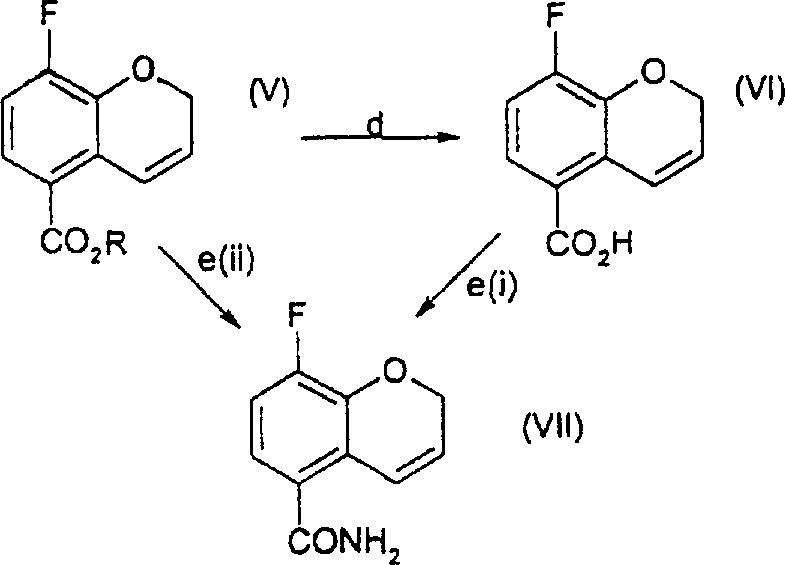
Following the formation of the benzopyran core, the pathway diverges to allow for flexible functionalization. As illustrated in the reaction scheme, Compound (V) can be hydrolyzed to the carboxylic acid (VI) or directly converted to the carboxamide (VII). The conversion to the amide is a pivotal step, as it sets the stage for the introduction of the nitrogen functionality at the 3-position. The subsequent nitration of Compound (VII) using iodine and nitrites introduces a nitro group selectively, which is then reduced to the amine. This sequence demonstrates a high degree of chemoselectivity, ensuring that the fluorine atom and the amide group remain intact while the double bond and nitro group are manipulated. The ability to perform these transformations with high yields, such as the 91% yield observed in the reduction of the nitro-alkene to the nitro-alkane, underscores the robustness of the chemical design.
How to Synthesize 3-N,N-dicyclobutylamino-8-fluoro-benzopyran Efficiently
The synthesis of the target molecule involves a logical progression of functional group interconversions that are well-suited for industrial application. The process initiates with the protection of the carboxylic acid, followed by the installation of the propargyl side chain which serves as the precursor for the pyran ring. The thermal cyclization step is the key rate-determining step, requiring careful temperature control to maximize the formation of the desired regioisomer. Once the core scaffold is established, the introduction of the amino group via nitration and reduction provides the handle for the final dialkylation. The detailed standardized synthesis steps, including specific solvent choices like acetone for alkylation and ethanol for resolution, are critical for reproducibility. For a comprehensive guide on executing these reactions with optimal parameters, please refer to the procedural breakdown below.
- Esterify 4-fluoro-3-hydroxybenzoic acid (II) using trialkyl orthoformate in anhydrous alcohol to form compound (III).
- Alkylate compound (III) with propargyl bromide in the presence of potassium carbonate to yield propargyl ether (IV).
- Perform thermal cyclization of compound (IV) at 210-230°C to form the benzopyran core (V), followed by hydrolysis or amidation to (VI) or (VII).
- Execute nitration to form compound (VIII), followed by selective reduction of the double bond to (IX) and subsequent reduction of the nitro group to amine (X).
- Resolve racemic amine (X) using L-(+)-tartaric acid to obtain R-(XI), followed by reductive alkylation with cyclobutanone to yield the final product (I).
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, the adoption of this novel synthetic route offers transformative benefits in terms of cost stability and raw material security. By relying on 4-fluoro-3-hydroxybenzoic acid as the starting material, manufacturers can bypass the volatile market for specialized fluorinating reagents and cryogenic services. The elimination of low-temperature lithiation steps means that production can be carried out in standard glass-lined steel reactors without the need for specialized cooling infrastructure, leading to substantial capital expenditure savings. Furthermore, the removal of chromatographic purification steps, which are notoriously difficult to scale and solvent-intensive, results in a drastic reduction in solvent consumption and waste disposal costs. This streamlined workflow translates directly into a more competitive pricing structure for the final intermediate, allowing pharmaceutical companies to optimize their bill of materials without compromising on quality.
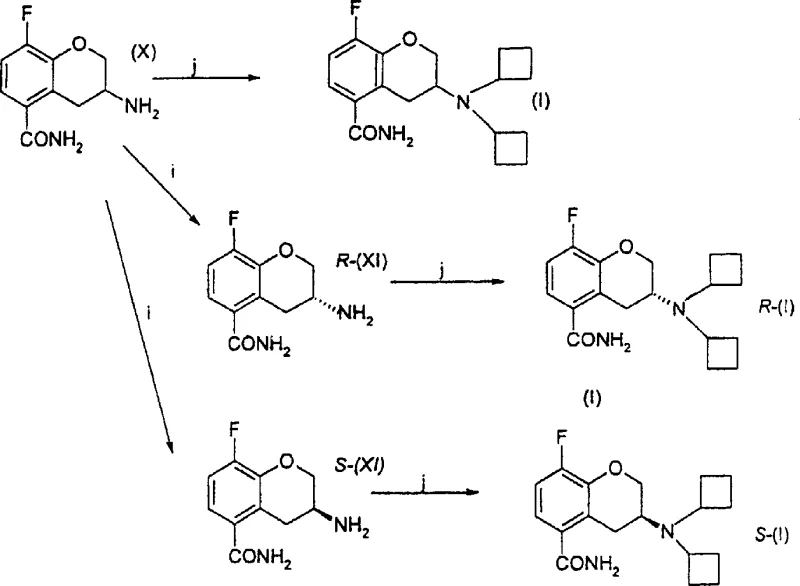
Enhanced supply chain reliability is another critical advantage conferred by this technology. The conventional route's dependence on multiple distinct chemical transformations increases the probability of batch failures and supply disruptions. In contrast, the new process features fewer unit operations and utilizes robust chemistry that is less sensitive to minor variations in reaction conditions. The final reductive alkylation step, shown in the diagram converting amines (X) and (XI) to the target dicyclobutylamine (I), employs common reagents like cyclobutanone and sodium cyanoborohydride, which are readily available in bulk quantities globally. This accessibility ensures that production schedules can be maintained consistently, reducing lead times for high-purity pharmaceutical intermediates. Additionally, the process allows for the isolation of stable intermediates like Compound (VII) and (X), which can be stockpiled as strategic reserves to buffer against upstream supply fluctuations, thereby enhancing overall supply chain resilience.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis. Understanding these details is essential for technical teams evaluating the feasibility of technology transfer. The answers provided are derived directly from the experimental data and claims within the patent documentation, ensuring accuracy and relevance for decision-makers.
Q: How does this process improve upon the method disclosed in WO 95/11891?
A: The conventional method requires late-stage fluorination via hazardous halogen-lithium exchange at low temperatures, generating difficult-to-separate by-products. This novel process utilizes a starting material with the fluorine atom pre-installed, eliminating the need for cryogenic lithiation and expensive fluorinating agents, thereby significantly simplifying the impurity profile and reducing operational hazards.
Q: What are the critical intermediates in this synthetic pathway?
A: Key intermediates include the propargyl ether derivative (IV), the cyclized benzopyran ester (V), the amide scaffold (VII), and the chiral amine precursors (R-XI and S-XI). These intermediates allow for flexible entry points into the synthesis and facilitate rigorous quality control at multiple stages before the final reductive alkylation.
Q: Is this manufacturing route suitable for large-scale commercial production?
A: Yes, the route is highly scalable. It replaces sensitive low-temperature organometallic steps with robust thermal cyclization reactions (210-230°C) and standard catalytic hydrogenations. The use of commercially available starting materials like 4-fluoro-3-hydroxybenzoic acid ensures supply chain continuity, while the avoidance of chromatographic purifications in later stages enhances throughput.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-N,N-dicyclobutylamino-8-fluoro-benzopyran Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthetic routes in the development of next-generation therapeutics. Our team of expert chemists has extensively analyzed the methodology presented in CN1259941A and possesses the technical capability to execute this pathway with precision. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move seamlessly from laboratory bench to full-scale manufacturing. Our facilities are equipped with state-of-the-art rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of intermediate delivered meets the highest international standards for pharmaceutical applications.
We invite you to collaborate with us to leverage this advanced technology for your drug development programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this route can optimize your manufacturing budget. Please contact us today to request specific COA data for our reference batches and to discuss route feasibility assessments for your upcoming projects. Let us be your partner in turning complex chemical challenges into commercial successes.