Scalable Four-Step Synthesis of Tert-Butyl-8-Oxylidene-2-Azaspiro Decane-2-Formic Acid Ester
Introduction to Advanced Spiro-Piperidine Scaffold Synthesis
The pharmaceutical industry continuously demands robust and scalable synthetic routes for complex heterocyclic scaffolds, particularly spiro-piperidine derivatives which serve as critical building blocks in modern drug discovery. Patent CN111518015A discloses a highly efficient preparation method for tert-butyl-8-oxylidene-2-azaspiro[4.5]decane-2-formic acid ester, a valuable intermediate characterized by its unique spirocyclic architecture. This technical breakthrough addresses significant bottlenecks in existing manufacturing protocols, specifically targeting the issues of high raw material costs, difficult reaction control, and inconvenient experimental operations that have historically plagued the synthesis of this compound class. By leveraging a rational four-step sequence starting from the inexpensive and commercially abundant 1,4-dioxaspiro[4.5]decane-8-ketone, this methodology offers a streamlined pathway that enhances both economic viability and operational safety for fine chemical manufacturers.
For R&D directors and process chemists, the significance of this patent lies in its ability to deliver high-purity intermediates through a sequence that avoids harsh conditions and exotic reagents. The strategic design of the synthetic route ensures that each transformation proceeds with high selectivity, minimizing the formation of difficult-to-remove impurities that often complicate downstream processing. As a reliable pharmaceutical intermediate supplier, understanding the nuances of such patented methodologies allows us to offer clients superior alternatives to legacy processes, ensuring that the supply chain for critical API precursors remains uninterrupted and cost-effective. The following analysis dissects the technical merits and commercial implications of this innovative synthesis strategy.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of spiro-piperidine ketones has been fraught with challenges that hinder their widespread adoption in large-scale manufacturing. Conventional routes often rely on expensive starting materials that are not readily available in bulk quantities, leading to prohibitive raw material costs that inflate the final price of the active pharmaceutical ingredient. Furthermore, many traditional methods suffer from low yields due to poor regioselectivity or the formation of stable byproducts that require extensive and wasteful purification steps. Reaction control is another major pain point, as older protocols may necessitate extreme temperatures or pressures that pose safety risks and require specialized reactor infrastructure, making batch production control difficult and inconsistent. These technical hurdles collectively render many existing synthesis processes unsuitable for amplification, forcing manufacturers to rely on inefficient small-scale batches that cannot meet the growing global demand for these complex heterocycles.
The Novel Approach
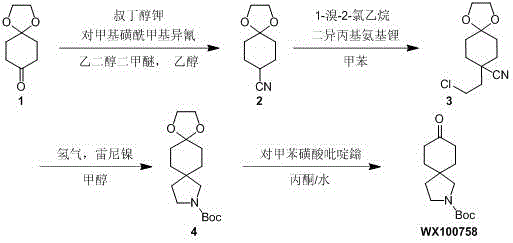
In stark contrast, the novel approach detailed in the patent utilizes a clever four-step cascade that transforms cheap and easily obtained 1,4-dioxaspiro[4.5]decane-8-ketone into the target spiro-piperidine ester with remarkable efficiency. The first step employs a TosMIC reaction to install a nitrile group, followed by a precise alkylation with 1-bromo-2-chloroethane to set up the nitrogen-containing ring precursor. The subsequent hydrogenative cyclization using Raney nickel is particularly elegant, simultaneously reducing the nitrile and closing the pyrrolidine ring in a single pot operation, which drastically simplifies the workflow. Finally, a mild acidic deprotection reveals the ketone functionality without compromising the integrity of the Boc-protected amine. This logical progression not only solves the technical problems of high cost and difficult control but also establishes a robust platform for cost reduction in pharmaceutical intermediate manufacturing by maximizing atom economy and minimizing waste generation.
Mechanistic Insights into Raney Nickel Catalyzed Cyclization
The third step of this synthesis represents the mechanistic cornerstone of the entire process, where the linear chloro-nitrile precursor is converted into the spiro-pyrrolidine scaffold. This transformation relies on the dual functionality of Raney nickel, which acts as a heterogeneous catalyst for the hydrogenation of the nitrile group to a primary amine intermediate. Under a hydrogen atmosphere of 50 Psi at 50°C in methanol, the nitrile is reduced in situ, generating a highly reactive amine species that immediately undergoes an intramolecular nucleophilic substitution with the pendant chloroethyl group. This tandem reduction-cyclization sequence is kinetically favored and thermodynamically driven by the formation of the stable five-membered pyrrolidine ring, effectively locking the spiro-architecture in place. The use of Raney nickel is advantageous here as it is a cost-effective catalyst that can be easily filtered off, avoiding the contamination issues associated with homogeneous transition metal catalysts and ensuring the final product meets stringent heavy metal specifications.
Following the cyclization, the crude amine is protected with di-tert-butyl dicarbonate (Boc2O) in the same reaction vessel, a strategy that prevents potential side reactions of the free amine during workup and storage. This one-pot procedure exemplifies process intensification, reducing solvent usage and processing time while maintaining high yield (80% in the optimized example). From an impurity control perspective, the mild reaction temperature of 50°C prevents the degradation of the sensitive acetal protecting group present on the cyclohexane ring, ensuring that the ketal remains intact until the dedicated deprotection step. This level of chemoselectivity is crucial for maintaining the structural integrity of the molecule and avoiding the formation of des-ketal impurities that would be difficult to separate, thereby guaranteeing a high-purity profile for the final pharmaceutical intermediate.

How to Synthesize Tert-Butyl-8-Oxylidene-2-Azaspiro Decane Efficiently
The synthesis of this complex spiro-piperidine derivative is achieved through a meticulously optimized four-step protocol that balances reaction kinetics with operational simplicity. The process begins with the cyanation of the starting ketone, followed by alkylation, cyclization, and finally deprotection, with each step designed to maximize yield and minimize purification burden. The detailed standardized synthetic steps below outline the specific reagents, stoichiometry, and conditions required to replicate this high-efficiency route in a GMP-compliant environment.
- React 1,4-dioxaspiro[4.5]decane-8-one with TosMIC and potassium tert-butoxide in glycol dimethyl ether/ethanol at 0-20°C to form the nitrile intermediate.
- Alkylate the nitrile intermediate with 1-bromo-2-chloroethane using LDA in toluene at 0-20°C to introduce the chloroethyl side chain.
- Perform hydrogenative cyclization using Raney nickel in methanol at 50°C and 50 Psi, followed by Boc protection to form the spiro-pyrrolidine ring.
- Deprotect the ketal group using pyridinium p-toluenesulfonate in acetone/water at 70°C to yield the final ketone product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers substantial strategic benefits that extend beyond simple chemistry. The primary advantage lies in the drastic simplification of the supply chain, as the starting material, 1,4-dioxaspiro[4.5]decane-8-ketone, is a commodity chemical that is cheap and easy to obtain in multi-ton quantities. This eliminates the reliance on bespoke, high-cost precursors that often create bottlenecks in the supply of critical intermediates. Additionally, the use of common industrial solvents such as toluene, methanol, and ethylene glycol dimethyl ether ensures that the process can be implemented in standard stainless steel reactors without the need for specialized glass-lined or Hastelloy equipment, further reducing capital expenditure and facilitating rapid technology transfer between manufacturing sites.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the elimination of expensive reagents and the optimization of step yields. By utilizing TosMIC and LDA, which are standard reagents in fine chemical synthesis, the process avoids the need for precious metal catalysts or complex chiral ligands that drive up costs. The high yield of the cyclization step (80%) and the efficient deprotection mean that less raw material is wasted per kilogram of final product, leading to significant cost savings in raw material procurement. Furthermore, the simplified workup procedures, which often involve simple extractions and crystallizations rather than complex chromatography on a large scale, reduce labor and utility costs associated with downstream processing.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route directly translates to improved supply chain continuity. Because the reaction conditions are mild (mostly 0-20°C for the early steps) and tolerant of minor variations, the risk of batch failure is significantly minimized compared to sensitive cryogenic or high-pressure processes. The availability of all reagents from multiple global suppliers mitigates the risk of single-source dependency, ensuring that production schedules can be maintained even during market fluctuations. This reliability is critical for long-term supply agreements with pharmaceutical partners who require guaranteed delivery of key intermediates to support their own clinical and commercial timelines.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, as demonstrated by the successful execution of the first step on a 500g scale in the patent examples, which serves as a strong proof-of-concept for multi-kilogram production. The use of Raney nickel, while requiring careful handling, is a well-established technology in the industry with established protocols for safe disposal and recycling, ensuring compliance with environmental regulations. The overall atom economy of the route is favorable, and the avoidance of halogenated solvents in the key cyclization step (using methanol instead) aligns with green chemistry principles, reducing the environmental footprint and simplifying waste treatment protocols for the manufacturing facility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this spiro-piperidine intermediate. These answers are derived directly from the experimental data and technical disclosures within the patent, providing a transparent view of the process capabilities and limitations for potential partners.
Q: What are the key advantages of this synthesis route compared to conventional methods?
A: This novel four-step process utilizes cheap and readily available starting materials like 1,4-dioxaspiro[4.5]decane-8-one, significantly lowering raw material costs. Furthermore, the reaction conditions are mild (0-20°C for early steps) and operationally convenient, solving the technical problems of difficult reaction control and low yields found in existing synthesis processes.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly demonstrates scalability with examples running on a 500g scale for the first step. The use of common solvents such as toluene, methanol, and ethylene glycol dimethyl ether, along with standard catalysts like Raney nickel, ensures that the process is easily adaptable for batch production control and industrial amplification without requiring exotic equipment.
Q: What is the overall yield and purity profile of the final product?
A: The process achieves a proper overall yield through optimized individual step yields, such as 74.76% for the initial cyanation and 80% for the cyclization step. The final product is obtained as a pure white solid after recrystallization and column purification, meeting stringent purity specifications required for pharmaceutical intermediate applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tert-Butyl-8-Oxylidene-2-Azaspiro Decane-2-Formic Acid Ester Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of your drug development programs. Our team of expert process chemists has thoroughly analyzed the patented route for tert-butyl-8-oxylidene-2-azaspiro[4.5]decane-2-formic acid ester and is fully prepared to implement this technology at commercial scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and precision. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of material we deliver meets the highest industry standards for pharmaceutical applications.
We invite you to collaborate with us to leverage this advanced synthesis technology for your next project. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis that details how switching to this optimized route can improve your project economics. We encourage you to contact us today to discuss your specific requirements,索取 specific COA data, and review our comprehensive route feasibility assessments to ensure a seamless integration of this intermediate into your supply chain.