Scalable Synthesis of Tetrahydrofuran Carboxylic Acid Intermediates for Oncology Applications
Scalable Synthesis of Tetrahydrofuran Carboxylic Acid Intermediates for Oncology Applications
The pharmaceutical landscape for prostate cancer treatment has been significantly shaped by potent androgen receptor inhibitors such as Enzalutamide (MDV3100) and ARN-509. As the demand for these life-saving therapeutics grows, the efficiency of their supply chains becomes paramount. Patent CN110669027B, filed in 2021, presents a critical technological advancement in the synthesis of key intermediates required for these drugs. Specifically, it discloses novel compounds of general formula 1, which are substituted 3-(4-methylcarbamoyl-3-fluorophenylamino)tetrahydrofuran-3-carboxylic acids and their esters. These molecules serve as pivotal building blocks in the construction of the complex spiro-hydantoin core found in next-generation anti-androgens. By addressing the synthetic bottlenecks associated with traditional methods, this intellectual property offers a pathway to more reliable pharmaceutical intermediate supplier networks and enhanced manufacturing economics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies for synthesizing androgen receptor inhibitors often relied on the direct interaction of 4-thioisocyanato-2-(trifluoromethyl)benzonitrile with amino-tetrahydrofuran derivatives. As illustrated in the reaction scheme below, conventional routes frequently necessitated harsh conditions, such as heating in dimethylformamide at 100°C for extended periods. More critically, these traditional pathways often resulted in racemic mixtures that required resolution via high-pressure liquid chromatography (HPLC) using chiral columns. This reliance on chiral chromatography represents a severe bottleneck in commercial manufacturing, as it drastically limits throughput, increases solvent consumption, and inflates the cost of goods sold (COGS). The difficulty in separating stereoisomers like A2 and A3 on a large scale has historically constrained the availability of high-purity active pharmaceutical ingredients (APIs).
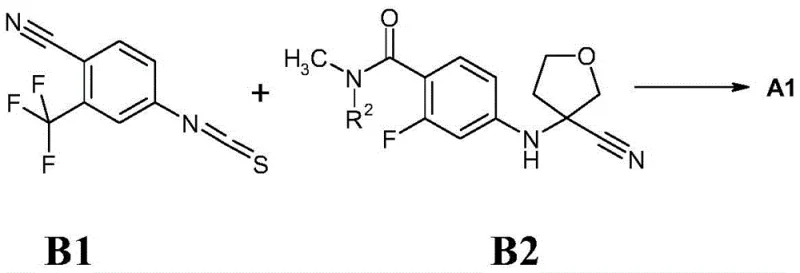
The Novel Approach
The innovation detailed in CN110669027B circumvents these historical challenges by introducing a modular synthetic strategy centered on stable, versatile intermediates of Formula 1. Instead of attempting to resolve the final complex molecule, the patent focuses on the efficient preparation of the tetrahydrofuran amino-acid scaffold itself. By utilizing specific esterification strategies (such as methyl or butyl esters) and optimized coupling conditions, the process achieves superior control over the molecular architecture before the final cyclization step. This strategic shift allows manufacturers to bypass the need for expensive chiral HPLC purification of the final drug substance, thereby enabling cost reduction in API manufacturing. The new approach leverages robust copper-catalyzed coupling reactions that are inherently more scalable and tolerant of industrial processing conditions compared to the fragile chromatographic separations of the past.
Mechanistic Insights into Copper-Catalyzed Amination and Cyclization
The core of this technological breakthrough lies in the formation of the C-N bond between the fluorinated benzamide moiety and the tetrahydrofuran ring. The patent describes a copper-catalyzed Ullmann-type coupling reaction where 4-bromo-2-fluoro-N-methylbenzamide reacts with 3-aminotetrahydrofuran-3-carboxylic acid derivatives. The mechanism likely involves the oxidative addition of the aryl bromide to a Copper(I) species, followed by coordination of the amine nucleophile and subsequent reductive elimination to forge the C-N linkage. The presence of ligands such as 2-acetylcyclohexanone and bases like potassium carbonate is crucial for stabilizing the catalytic cycle and neutralizing the hydrogen bromide byproduct. This mechanistic understanding is vital for R&D directors aiming to optimize reaction kinetics and minimize impurity profiles, ensuring that the resulting intermediate meets stringent purity specifications required for oncology drugs.
Following the formation of the intermediate, the final assembly of the spiro-hydantoin core involves a nucleophilic attack of the secondary amine on the isothiocyanate group of the trifluoromethyl-benzonitrile partner. The patent highlights that performing this reaction in a specific solvent system—comprising dimethyl sulfoxide and ethyl acetate—significantly enhances the reaction outcome. This solvent mixture likely optimizes the solubility of both the polar intermediate and the less polar isothiocyanate, facilitating effective molecular collisions. Furthermore, the reaction conditions promote the intramolecular cyclization to form the thioxo-diazaspiro ring system with high regioselectivity. By controlling the stoichiometry and temperature, the process minimizes the formation of urea byproducts or hydrolysis of the nitrile group, resulting in a cleaner crude profile that simplifies downstream processing.
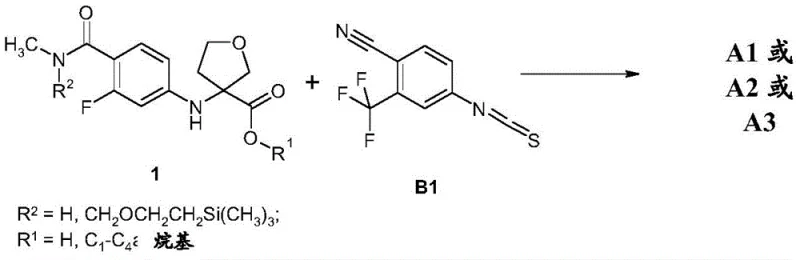
How to Synthesize Tetrahydrofuran Carboxylic Acid Efficiently
The preparation of these high-value intermediates requires precise adherence to the optimized protocols disclosed in the patent to ensure maximum yield and stereochemical integrity. The process begins with the selection of appropriate protecting groups and ester variants to modulate solubility and reactivity during the coupling phase. Detailed operational parameters, including specific molar ratios of catalysts and bases, are essential for reproducing the reported success rates. For process chemists looking to implement this technology, the following guide outlines the critical stages of the synthesis, emphasizing the transition from raw materials to the purified intermediate ready for final cyclization.
- React 4-bromo-2-fluoro-N-methylbenzamide with 3-aminotetrahydrofuran-3-carboxylic acid or its ester using Copper(I) iodide and potassium carbonate in DMF/Water.
- Isolate the intermediate ester or acid, ensuring high purity through acidification and filtration techniques described in the patent examples.
- Couple the purified intermediate with 4-isothiocyanato-2-trifluoromethylbenzonitrile in a DMSO/Ethyl Acetate mixture to form the final spiro-hydantoin structure.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of the synthetic route described in CN110669027B translates into tangible strategic benefits beyond mere chemical elegance. The primary advantage is the drastic simplification of the purification train. By eliminating the dependency on preparative chiral chromatography for the final isolation of the API, manufacturers can significantly reduce capital expenditure on specialized equipment and lower the operational costs associated with high-purity solvent usage. This streamlining of the process directly contributes to substantial cost savings in pharmaceutical intermediate production, making the final therapy more accessible. Additionally, the use of commodity chemicals such as copper iodide and potassium carbonate ensures that the supply chain remains resilient against fluctuations in the availability of exotic reagents.
- Cost Reduction in Manufacturing: The elimination of chiral HPLC separation steps removes one of the most expensive unit operations in small molecule drug manufacturing. This process intensification allows for higher batch sizes and reduced cycle times, leading to a more favorable economic model for producing complex spiro-hydantoin structures. Furthermore, the improved yields reported in the patent, reaching up to 70 percent for the final coupling step, mean that less raw material is wasted, further driving down the variable costs per kilogram of produced intermediate.
- Enhanced Supply Chain Reliability: The synthetic route relies on robust, well-understood chemical transformations that are less prone to failure than delicate chromatographic resolutions. This reliability ensures consistent delivery schedules for downstream API manufacturers, reducing the risk of drug shortages. The ability to produce the intermediate as a stable ester (such as the methyl or butyl ester) also offers flexibility in logistics, allowing for safer storage and transportation before the final deprotection and cyclization steps are performed at the API site.
- Scalability and Environmental Compliance: The shift towards a solution-based crystallization and filtration workflow, as opposed to chromatography, aligns better with green chemistry principles by reducing solvent waste intensity. The process is designed to be scalable from laboratory benchtop to multi-ton commercial production without fundamental changes to the chemistry. This scalability ensures that as market demand for androgen receptor inhibitors grows, the supply infrastructure can expand seamlessly to meet global needs without compromising on environmental standards or regulatory compliance.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These insights are derived directly from the experimental data and claims within the patent documentation, providing clarity for technical teams evaluating the feasibility of this route for their specific portfolio needs.
Q: How does this patent improve upon the synthesis of Enzalutamide intermediates?
A: The patent introduces a novel intermediate (Formula 1) that allows for the introduction of chirality earlier in the synthesis or avoids difficult chiral separations required in conventional routes, significantly simplifying the process.
Q: What are the optimal reaction conditions for the final cyclization step?
A: The patent specifies that best results are achieved using a mixture of dimethyl sulfoxide and ethyl acetate (1:2 ratio) under an argon atmosphere at elevated temperatures, yielding up to 70% product.
Q: Can this process be scaled for commercial API production?
A: Yes, the use of standard reagents like Copper(I) iodide and Potassium Carbonate, along with the avoidance of preparative chiral HPLC for the final product, makes this route highly suitable for multi-kilogram commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tetrahydrofuran Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust intermediate supply chains in the development of life-saving oncology therapies. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from patent literature to industrial reality is seamless. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs and state-of-the-art analytical capabilities. Our expertise in copper-catalyzed couplings and heterocyclic chemistry positions us as an ideal partner for executing the complex synthesis of tetrahydrofuran-based androgen receptor inhibitor precursors.
We invite global pharmaceutical partners to engage with our technical procurement team to discuss how this innovative synthetic route can be integrated into your supply chain. By leveraging our manufacturing capabilities, you can secure a stable source of critical materials while optimizing your overall production costs. Please contact us today to request a Customized Cost-Saving Analysis, specific COA data for our reference standards, and comprehensive route feasibility assessments tailored to your project timelines.