Advanced Manufacturing of 2-Chloro-4-(methoxymethyl)pyrimidine for Global Pharmaceutical Supply Chains
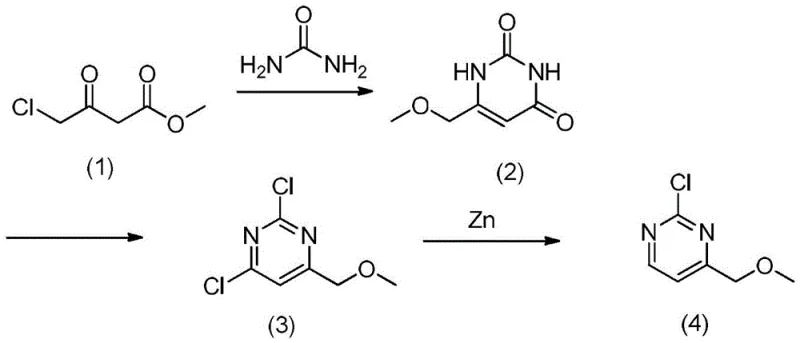
The pharmaceutical industry constantly seeks robust and scalable synthetic routes for critical heterocyclic intermediates, and the recent disclosure of patent CN115947692A offers a significant advancement in the production of 2-chloro-4-(methoxymethyl)pyrimidine. This specific pyrimidine derivative serves as a vital building block for constructing biologically active compounds, particularly in the realm of medicinal chemistry where the pyrimidine ring skeleton is ubiquitous. The traditional landscape for synthesizing such substituted pyrimidines has often been fraught with challenges regarding impurity profiles and process control, necessitating a re-evaluation of manufacturing strategies. The methodology outlined in this patent introduces a novel three-step sequence that begins with the readily available methyl 4-chloroacetoacetate, proceeding through a urea-mediated cyclization, followed by chlorination, and concluding with a selective dechlorination. This approach is not merely an academic exercise but represents a tangible solution for reliable pharmaceutical intermediate supplier networks aiming to secure high-purity materials. By fundamentally redesigning the reaction pathway, the inventors have addressed long-standing issues related to by-product formation, thereby enhancing the overall quality of the final API intermediate. For R&D directors and procurement specialists, understanding the nuances of this new route is essential for evaluating its potential integration into existing supply chains and for assessing the feasibility of cost reduction in pharmaceutical intermediate manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-chloro-4-(methoxymethyl)pyrimidine and related analogs has been characterized by significant technical hurdles that impede efficient commercial production. Conventional methods often suffer from poor selectivity, leading to complex mixtures of regioisomers and side products that are notoriously difficult to separate during the purification phase. These impurities can persist through multiple downstream processing steps, ultimately compromising the purity specifications required for pharmaceutical applications. Furthermore, many traditional routes rely on harsh reaction conditions or expensive reagents that drive up the cost of goods sold and introduce safety hazards in a plant environment. The literature indicates that public reports on the synthesis of this specific compound are insufficient, suggesting that existing methods lack the robustness needed for consistent large-scale manufacturing. Process reproducibility is another critical pain point; slight variations in temperature or reagent addition in older protocols can lead to drastic drops in yield or the formation of intractable tars. For supply chain heads, these inconsistencies translate into unpredictable lead times and potential disruptions in the availability of high-purity pharmaceutical intermediates. The inability to effectively remove by-products before the final isolation step often necessitates extensive chromatographic purification, which is neither economically nor environmentally sustainable for kilogram-to-ton scale operations.
The Novel Approach
In stark contrast to these legacy challenges, the novel approach detailed in the patent data presents a streamlined and chemically elegant solution that prioritizes both yield and purity. The strategy employs a distinct sequence where the pyrimidine ring is constructed early using a Lewis acid-catalyzed cyclization, ensuring a stable core before subsequent functionalization. A key innovation lies in the timing of impurity removal; the process is designed such that major by-products are eliminated prior to the final synthetic step, drastically simplifying the workup procedure. This foresight in process design means that the final dechlorination step proceeds with a cleaner substrate, minimizing the risk of contaminating the final product with hard-to-remove impurities. The use of common industrial solvents like toluene and methanol, combined with moderate heating requirements ranging from 70°C to 90°C, underscores the practicality of this method for commercial scale-up of complex pharmaceutical intermediates. The reaction conditions are described as easy to control, which is a paramount consideration for plant operators managing large reactors. By avoiding the generation of impurities through route design rather than relying solely on purification, this method offers a substantial cost savings potential and a more reliable supply of the target molecule. The successful demonstration of this route at the kilogram reaction scale provides concrete evidence of its viability for industrial adoption.
Mechanistic Insights into Lewis Acid-Catalyzed Cyclization and Selective Reduction
The chemical elegance of this synthesis is rooted in the precise control of reactivity at each stage, beginning with the formation of the pyrimidine ring. The first step involves the condensation of methyl 4-chloroacetoacetate with urea, a reaction facilitated by the presence of a Lewis acid catalyst such as triphenylboron or boron trifluoride. This catalytic activation is crucial for promoting the cyclization under relatively mild thermal conditions, typically between 70°C and 90°C, which helps to preserve the integrity of the sensitive ester and chloromethyl functionalities. Following the initial ring closure, the intermediate undergoes a methoxylation step using sodium methoxide, which installs the methoxymethyl group essential for the final structure. The subsequent chlorination step utilizes phosphorus pentachloride and polyphosphoric acid to introduce chlorine atoms at the 2 and 4 positions of the pyrimidine ring, creating a dichloro-intermediate that is primed for selective reduction. The final transformation is particularly noteworthy, employing zinc powder in the presence of trimethylchlorosilane and an acid to selectively remove one of the chlorine atoms. This chemoselective dechlorination is the key to obtaining the 2-chloro-4-(methoxymethyl) substitution pattern without over-reduction or loss of the methoxy group.
From an impurity control perspective, the mechanism offers distinct advantages over random or non-optimized pathways. The sequential nature of the synthesis allows for the isolation and purification of intermediates, specifically the removal of by-products generated during the chlorination phase before they can interfere with the final reduction. This intermediate purification strategy is critical for maintaining a clean impurity profile, as it prevents the carryover of phosphorus-containing residues or over-chlorinated species into the final product. The use of zinc powder as a reducing agent is a well-established, cost-effective method that avoids the need for expensive transition metal catalysts which often require rigorous removal to meet heavy metal specifications in pharmaceuticals. The reaction environment in the final step, utilizing methanol and acid, ensures that the zinc surface remains active for the reduction while keeping the organic intermediates in solution. This mechanistic understanding assures R&D directors that the process is not only feasible but also robust against common failure modes associated with heterocyclic synthesis. The ability to tune the molar ratios of reagents, such as the zinc to intermediate ratio of 1:2-4, provides an additional handle for optimizing yield and minimizing side reactions, further enhancing the reliability of the manufacturing process.
How to Synthesize 2-Chloro-4-(methoxymethyl)pyrimidine Efficiently
Implementing this synthesis in a production environment requires a clear understanding of the operational parameters and safety considerations associated with each step. The process begins with the careful handling of Lewis acids and urea to form the initial ring system, followed by a controlled chlorination that requires management of exotherms and acidic by-products. The final reduction step necessitates the safe handling of zinc powder and the management of hydrogen evolution, which is typical for metal-acid reductions. Detailed standard operating procedures (SOPs) would specify the exact addition rates, temperature ramps, and quenching protocols to ensure consistent batch-to-batch quality. The patent data provides a solid foundation for these procedures, outlining specific solvent choices like toluene for the chlorination and methanol for the reduction, which are familiar to most chemical manufacturing facilities. For technical teams looking to adopt this route, the focus should be on optimizing the workup procedures to maximize the removal of inorganic salts and metal residues. The simplicity of the post-treatment, often involving filtration and extraction, suggests that the process is amenable to automation and continuous processing technologies. Below is the structured guide for the synthesis steps based on the patent specifications.
- Cyclization of methyl 4-chloroacetoacetate with urea using a Lewis acid catalyst to form the pyrimidine ring intermediate.
- Chlorination of the intermediate using phosphorus pentachloride and polyphosphoric acid in an organic solvent.
- Selective dechlorination using zinc powder and trimethylchlorosilane to yield the final high-purity product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this new synthetic route translates into tangible strategic advantages that go beyond simple chemistry. The primary benefit lies in the significant cost reduction in pharmaceutical intermediate manufacturing driven by the simplification of the purification process. By designing a route that avoids the generation of difficult impurities, the need for expensive and time-consuming chromatographic separations is eliminated or drastically reduced. This directly impacts the cost of goods, allowing for more competitive pricing without sacrificing margin. Furthermore, the use of readily available raw materials such as methyl 4-chloroacetoacetate, urea, and zinc powder ensures that the supply chain is not dependent on exotic or single-source reagents that could pose availability risks. The robustness of the reaction conditions, which operate at moderate temperatures and utilize common solvents, reduces the energy consumption and specialized equipment requirements, further contributing to overall cost efficiency. For supply chain planners, the proven scalability to the kilogram level indicates a low risk of failure during technology transfer to larger production vessels. This reliability is crucial for maintaining continuous supply to downstream API manufacturers, reducing the likelihood of stockouts or production delays. The process's environmental profile is also improved by minimizing waste generation and avoiding the use of heavy metal catalysts, which aligns with increasingly stringent global environmental regulations and sustainability goals.
- Cost Reduction in Manufacturing: The elimination of complex purification steps and the use of cost-effective reagents like zinc powder significantly lower the operational expenditure associated with producing this intermediate. By removing by-products early in the sequence, the process avoids the yield losses typically associated with late-stage purification, thereby improving the overall material efficiency. This logical deduction of cost savings is based on the mechanistic advantage of the route rather than arbitrary financial projections, ensuring a realistic assessment of economic benefits. The avoidance of expensive transition metal catalysts also removes the need for costly metal scavenging steps, which are often a hidden cost in pharmaceutical manufacturing. Consequently, the total cost of production is optimized, making the final product more attractive for high-volume applications.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals and standard solvents means that the supply chain is resilient to market fluctuations that might affect specialized reagents. The simplicity of the operation reduces the dependency on highly specialized operator skills, allowing for more flexible manufacturing scheduling and easier replication across different sites. This redundancy in manufacturing capability is a key factor in reducing lead time for high-purity pharmaceutical intermediates, as production can be ramped up quickly in response to demand spikes. The proven reproducibility of the process ensures that quality remains consistent regardless of the batch size, which is essential for maintaining regulatory compliance and customer trust. Supply chain heads can therefore plan with greater confidence, knowing that the technical risks associated with scale-up have been mitigated by the robust design of the synthesis.
- Scalability and Environmental Compliance: The process is explicitly designed for industrial scale-up, with conditions that are safe and manageable in large reactors. The removal of by-products before the final step simplifies the waste stream, making treatment and disposal more straightforward and cost-effective. This aligns with green chemistry principles by reducing the E-factor of the process, which is a critical metric for environmental compliance in the fine chemical industry. The use of zinc and common acids generates waste that is easier to handle compared to heavy metal waste, reducing the environmental footprint of the manufacturing site. Scalability is further supported by the exothermic nature of the reactions being manageable within standard cooling capacities, ensuring that safety is maintained even at the 100 MT scale. This combination of scalability and environmental stewardship makes the route highly attractive for long-term commercial partnerships.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of 2-chloro-4-(methoxymethyl)pyrimidine. These answers are derived directly from the technical specifications and beneficial effects described in the patent literature, providing a reliable source of information for decision-makers. Understanding these details is crucial for evaluating the fit of this intermediate within your specific drug development pipeline or manufacturing portfolio. The responses cover aspects of purity, scalability, and process safety, which are the primary concerns for R&D and operations teams. By clarifying these points, we aim to facilitate a smoother evaluation process and accelerate the integration of this technology into your supply chain.
Q: What are the key advantages of the new synthesis route for 2-chloro-4-(methoxymethyl)pyrimidine?
A: The new route described in patent CN115947692A avoids the generation of difficult-to-remove impurities common in conventional methods. It allows for the removal of by-products before the final step, simplifying post-treatment and ensuring high product quality suitable for pharmaceutical applications.
Q: Is this synthesis method scalable for industrial production?
A: Yes, the patent explicitly states that the process has good reproducibility and can be successfully scaled up to the kilogram level. The use of common solvents like toluene and methanol, along with stable reaction conditions (70-90°C), supports commercial scale-up.
Q: What are the critical reagents used in the final reduction step?
A: The final step involves the reduction of the dichloro-intermediate using zinc powder as the reducing agent, in the presence of trimethylchlorosilane and an acid (such as hydrochloric acid or glacial acetic acid) in methanol.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Chloro-4-(methoxymethyl)pyrimidine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of pharmaceutical development and commercialization. Our team of experts possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition seamlessly from the lab to the plant. We are committed to delivering products that meet stringent purity specifications through our rigorous QC labs, which employ state-of-the-art analytical techniques to verify every batch. Our capability to implement the advanced synthesis route described in patent CN115947692A allows us to offer a superior product with a cleaner impurity profile and a more competitive cost structure. We understand that consistency is key, and our manufacturing processes are designed to minimize variability, providing you with a reliable partner for your long-term supply needs. Whether you require custom synthesis or standard catalog items, our infrastructure is built to support your growth and innovation.
We invite you to engage with our technical procurement team to discuss how we can optimize your supply chain for this critical intermediate. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our manufacturing efficiencies can translate into value for your organization. We encourage you to reach out for specific COA data and route feasibility assessments to verify our capabilities against your specific requirements. Our goal is to establish a transparent and collaborative partnership that drives mutual success in the competitive pharmaceutical market. Let us handle the complexities of chemical manufacturing so you can focus on delivering life-saving therapies to patients worldwide.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →