Scalable Synthesis of High-Purity Silodosin Dehydrogenated Impurity for Global Pharmaceutical Quality Control
The pharmaceutical industry relies heavily on precise impurity profiling to ensure the safety and efficacy of active pharmaceutical ingredients (APIs), particularly for complex molecules like Silodosin, a selective alpha-1A adrenoceptor antagonist used in treating benign prostatic hyperplasia. Patent CN111116448B introduces a robust preparation method for a critical indole derivative, specifically the dehydrogenated impurity of Silodosin, which serves as an essential reference standard for quality control laboratories. This technical breakthrough addresses the longstanding challenge of obtaining high-purity samples of this specific structural analog, which is notoriously difficult to isolate from crude reaction mixtures due to its physical properties. By establishing a dedicated synthetic route rather than relying on extraction from process waste, manufacturers can secure a reliable supply of calibration standards necessary for rigorous HPLC analysis and regulatory compliance. The methodology outlined in this patent demonstrates a sophisticated understanding of heterocyclic chemistry, leveraging protective group strategies and selective oxidation to achieve purity levels exceeding 95.0 percent, thereby setting a new benchmark for impurity standard preparation in the fine chemical sector.
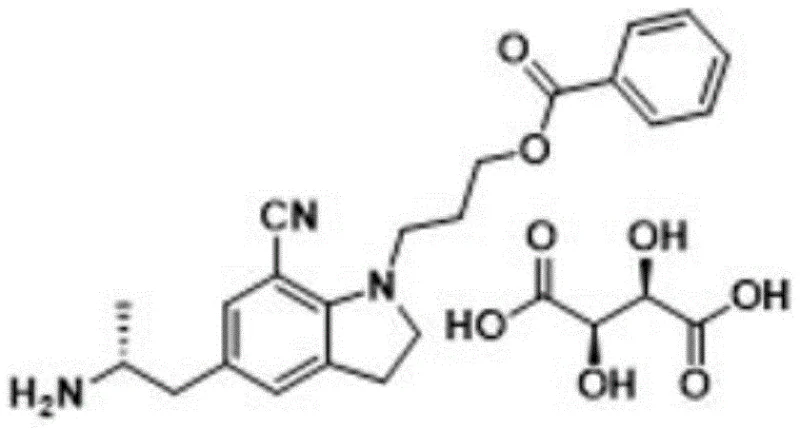
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the acquisition of specific process-related impurities for drugs like Silodosin has been fraught with significant technical hurdles, primarily stemming from the reliance on isolating these byproducts from the main synthesis stream. In conventional Silodosin manufacturing, the dehydrogenated impurity often co-elutes with the desired product or exists as a viscous oil that resists crystallization, making it extremely difficult to purify to the high standards required for analytical reference materials. The structural similarity between the saturated dihydroindole backbone of the drug and the aromatic indole backbone of the impurity means that standard purification techniques like recrystallization are often ineffective, leading to low recovery rates and inconsistent purity profiles. Furthermore, the oily nature of the free base form of these impurities complicates handling, weighing, and long-term storage, introducing variability into quality control assays that can compromise the validity of batch release testing. This lack of a dedicated, high-yield synthesis for the impurity itself creates a bottleneck for pharmaceutical companies striving to meet stringent regulatory requirements for impurity identification and quantification.
The Novel Approach
The innovative strategy presented in the patent data circumvents these isolation issues by synthesizing the target impurity directly through a controlled, multi-step chemical transformation designed to maximize yield and purity from the outset. Instead of attempting to separate the impurity from the API, this approach starts with a protected precursor and deliberately induces aromatization of the indole ring system using specific oxidizing agents. By employing an amino protection step prior to oxidation, the synthesis prevents unwanted side reactions at the amine functionality, ensuring that the oxidative dehydrogenation occurs selectively at the pyrrole ring. The subsequent deprotection and salt formation steps are engineered to convert the typically oily free base into a stable, crystalline solid salt, such as the oxalate or tartrate, which is far easier to handle and characterize. This deliberate construction of the molecule allows for the production of the impurity standard with a calibration content of greater than 95.0 percent, providing a reliable tool for qualitative and quantitative research that was previously unavailable through traditional extraction methods.
Mechanistic Insights into Oxidative Dehydrogenation and Protection Strategies
The core chemical transformation in this synthesis revolves around the oxidative dehydrogenation of the 2,3-dihydro-1H-indole scaffold to form the fully aromatic 1H-indole system, a reaction that requires precise control to avoid over-oxidation or degradation of sensitive functional groups. The mechanism typically involves the use of quinone-based oxidants like 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) or heterogeneous oxidants like manganese dioxide, which facilitate the removal of two hydrogen atoms from the saturated five-membered ring. This aromatization process is thermodynamically favorable due to the gain in resonance energy associated with the formation of the aromatic indole nucleus, but kinetically it requires careful selection of reaction conditions to ensure selectivity. The presence of the cyano group at the 7-position and the benzoyloxypropyl chain at the 1-position adds steric and electronic complexity, necessitating a solvent system that can dissolve the intermediate while maintaining the stability of the oxidant. The patent specifies solvents such as ethyl acetate or dichloromethane, which provide the appropriate polarity to support the reaction kinetics without promoting hydrolysis of the ester or nitrile functionalities.
Equally critical to the success of this route is the strategic implementation of amino protection and deprotection cycles to manage the reactivity of the primary amine side chain. During the oxidation step, the free amine is susceptible to nucleophilic attack or oxidation itself, which could lead to the formation of imines, nitriles, or polymeric byproducts that would contaminate the final product. By masking the amine with a tert-butoxycarbonyl (Boc) group or similar protecting agents in the initial step, the synthesis effectively renders the nitrogen atom inert to the oxidative conditions. Following the successful formation of the indole ring, the protecting group is removed under acidic conditions, such as treatment with trifluoroacetic acid or hydrochloric acid, regenerating the free amine without disturbing the newly formed aromatic system. This orthogonal protection strategy ensures that the final deprotection yields the desired amine cleanly, which is then immediately converted into a stable salt form to lock in the purity and prevent degradation during storage and transport.
How to Synthesize 5-[(2R)-2-aminopropyl]-1-[3-(benzoyloxy)propyl]-7-cyano-1H-indole Efficiently
The synthesis of this high-value pharmaceutical intermediate requires a disciplined approach to reaction monitoring and workup procedures to maintain the integrity of the chiral center and the functional groups throughout the four-step sequence. Operators must pay close attention to the stoichiometry of the protecting agent and the oxidant, as deviations can lead to incomplete conversion or the formation of difficult-to-remove side products. The initial protection step sets the stage for the entire synthesis, requiring thorough mixing and temperature control to ensure uniform derivatization of the starting tartrate salt. Subsequent oxidation demands careful quenching and filtration to remove reduced oxidant byproducts, while the final salt formation step relies on precise pH control and cooling rates to induce the crystallization of the target oxalate or tartrate salt. For a detailed breakdown of the specific operational parameters, reagent grades, and safety precautions required for this process, please refer to the standardized synthesis guide below.
- Perform amino protection on the tartrate precursor using a protecting agent like di-tert-butyl dicarbonate in the presence of a base such as triethylamine.
- Execute oxidative dehydrogenation using oxidants like DDQ or manganese dioxide to convert the dihydroindole core into the aromatic indole structure.
- Conduct acidic deprotection to remove the amino protecting group, followed by salt formation with oxalic or tartaric acid to isolate the final solid product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers substantial strategic benefits by transforming a hard-to-source analytical standard into a commercially viable product with consistent quality attributes. The ability to produce the impurity as a stable solid salt rather than an unstable oil significantly reduces the risks associated with storage, shipping, and handling, thereby minimizing waste and ensuring that the material remains fit for purpose over extended periods. This stability translates directly into supply chain reliability, as inventory can be held with confidence that the purity specifications will not degrade over time, reducing the need for frequent re-testing or replacement of expired standards. Furthermore, the high purity achieved through this dedicated synthesis eliminates the need for costly and time-consuming preparative chromatography purification steps that are typically required when isolating impurities from crude API batches. This streamlined production process lowers the overall cost of goods sold for the impurity standard, allowing suppliers to offer more competitive pricing while maintaining healthy margins.
- Cost Reduction in Manufacturing: The elimination of complex purification workflows, such as repeated column chromatography or preparative HPLC, drastically simplifies the manufacturing process and reduces the consumption of expensive solvents and stationary phases. By designing the synthesis to yield a crystallizable solid directly, the process avoids the yield losses typically associated with isolating oily residues, leading to a more efficient use of raw materials. Additionally, the use of common, commercially available oxidants and protecting agents ensures that the input costs remain stable and predictable, shielding the supply chain from volatility associated with exotic or specialized reagents. This operational efficiency allows for a significant reduction in the overall production cost, making high-purity impurity standards more accessible to pharmaceutical quality control departments without compromising on the rigorous specifications required for regulatory compliance.
- Enhanced Supply Chain Reliability: The robustness of the four-step synthetic route ensures a consistent and dependable supply of the Silodosin impurity standard, mitigating the risk of shortages that can halt quality control operations in API manufacturing facilities. Because the process does not rely on the availability of large volumes of Silodosin crude product for extraction, the production of the impurity can be scaled independently based on market demand for analytical standards. This decoupling from the main API production schedule provides greater flexibility in planning and inventory management, ensuring that customers can secure the materials they need for method validation and batch release testing without delay. The high yield and reproducibility of the method further contribute to supply security, as manufacturers can confidently forecast production output and commit to long-term supply agreements with key pharmaceutical partners.
- Scalability and Environmental Compliance: The synthesis utilizes standard organic solvents and reagents that are well-understood in terms of waste management and environmental impact, facilitating easier regulatory approval for commercial-scale production. The ability to crystallize the final product reduces the volume of solvent waste generated compared to processes that rely on distillation or chromatography for purification of oils, aligning with green chemistry principles and corporate sustainability goals. Moreover, the solid nature of the final salt form simplifies packaging and disposal of unused material, reducing the environmental footprint associated with the lifecycle of the chemical. This scalability ensures that the supply can easily grow from laboratory gram quantities to multi-kilogram commercial batches to support the expanding global demand for Silodosin generics and the accompanying need for rigorous quality control standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this specific indole derivative, drawing directly from the experimental data and claims within the patent documentation. Understanding these details is crucial for R&D teams evaluating the suitability of this material for their analytical methods and for procurement specialists assessing the vendor's technical capability. The answers provided reflect the specific advantages of the oxidative dehydrogenation pathway and the salt formation strategy that distinguishes this product from lower-purity alternatives available in the market.
Q: Why is the dehydrogenated impurity of Silodosin difficult to purify in conventional processes?
A: In standard Silodosin manufacturing, the dehydrogenated byproduct often exists as an oily substance with a structure highly similar to the active pharmaceutical ingredient, making chromatographic separation challenging and resulting in low purity standards.
Q: What is the advantage of using the oxidative dehydrogenation method described in CN111116448B?
A: This method intentionally synthesizes the impurity through a controlled protection-oxidation-deprotection sequence, allowing for the isolation of the target compound as a stable solid salt with purity exceeding 95%, suitable for use as a reference standard.
Q: Which oxidants are preferred for the aromatization step in this synthesis?
A: The patent highlights 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) and manganese dioxide as effective oxidants for converting the 2,3-dihydro-1H-indole intermediate into the fully aromatic 1H-indole derivative under mild conditions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-[(2R)-2-aminopropyl]-1-[3-(benzoyloxy)propyl]-7-cyano-1H-indole Supplier
As a premier CDMO and fine chemical manufacturer, NINGBO INNO PHARMCHEM possesses the technical expertise and infrastructure to replicate and optimize the synthesis of complex pharmaceutical intermediates like the Silodosin dehydrogenated impurity described in CN111116448B. Our facility is equipped with state-of-the-art reactors and purification systems capable of handling the specific oxidative and protection chemistries required for this route, ensuring that every batch meets the stringent purity specifications demanded by global regulatory bodies. We have extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, allowing us to seamlessly transition this synthesis from laboratory development to full-scale manufacturing to support your growing needs. Our rigorous QC labs employ advanced analytical techniques, including HPLC, NMR, and Mass Spectrometry, to verify the identity and purity of every lot, guaranteeing that the material you receive is fit for use as a critical reference standard in your quality control workflows.
We invite you to collaborate with our technical team to explore how this high-purity intermediate can enhance your Silodosin quality control protocols and streamline your regulatory compliance efforts. By partnering with us, you gain access to a Customized Cost-Saving Analysis that evaluates the economic benefits of sourcing this material from our optimized supply chain versus internal production or alternative vendors. We encourage you to contact our technical procurement team today to request specific COA data for recent batches and to discuss route feasibility assessments for any related indole derivatives you may require for your pharmaceutical development pipeline.