Advanced Asymmetric Synthesis of 2-Fluoro-L-ristosamine for Commercial Pharmaceutical Manufacturing
The pharmaceutical industry continuously seeks robust synthetic routes for complex carbohydrate derivatives, particularly fluorinated sugars that serve as critical building blocks for next-generation therapeutics. Patent CN109503681B introduces a groundbreaking asymmetric synthesis method for 2-Fluoro-L-ristosamine, a specialized 2,6-dideoxy sugar analogue with significant potential in the development of antibacterial and antitumor agents. This technology addresses the longstanding challenges associated with introducing fluorine atoms into specific positions of sugar moieties without compromising stereochemical integrity. By leveraging a sequence of coupling, Sharpless epoxidation, and organocatalytic fluorination, the disclosed method offers a pathway to high-purity intermediates that are essential for the manufacture of potent glycoside antibiotics. For R&D directors and procurement specialists, understanding this patent is crucial as it represents a shift towards more sustainable and economically viable manufacturing processes for high-value pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fluorinated deoxysugars has been plagued by significant technical and economic hurdles that hinder large-scale commercial adoption. Traditional strategies often rely on the use of hazardous fluorinating agents such as xenon difluoride, which poses severe safety risks and requires specialized handling infrastructure, thereby inflating operational costs. Furthermore, earlier methodologies involving the synthesis of 2,3-epoxy sugars followed by nucleophilic ring opening frequently suffer from poor regioselectivity and competing side reactions like Payne rearrangement. These inefficiencies typically result in suboptimal yields, often ranging between 57% and 60%, necessitating extensive purification steps that erode profit margins. The reliance on expensive protecting group strategies and harsh reaction conditions further complicates the supply chain, making it difficult to secure a consistent supply of high-quality intermediates for drug development pipelines.
The Novel Approach
In stark contrast to these legacy techniques, the novel approach detailed in the patent utilizes a convergent strategy starting from simple, commercially abundant raw materials such as 3-butyn-2-ol and 3-bromopropene. This method eliminates the need for toxic heavy metal catalysts and dangerous fluorinating gases, replacing them with safer, organocatalytic systems that operate under mild conditions. The process is designed with step economy in mind, streamlining the transformation from acyclic precursors to the cyclic sugar framework through a logical sequence of reduction, epoxidation, and oxidative cleavage. By avoiding the pitfalls of low-yielding ring-opening reactions and utilizing highly selective enzymatic-mimetic catalysis, this new route drastically simplifies the post-reaction workup. This technological leap not only enhances the overall atom economy but also ensures a more predictable and stable manufacturing process, which is vital for maintaining continuity in the global supply of critical antibiotic intermediates.
Mechanistic Insights into Organocatalytic Asymmetric Fluorination
The core innovation of this synthesis lies in its sophisticated control over stereochemistry, achieved primarily through Sharpless asymmetric epoxidation and subsequent organocatalytic fluorination. The process begins with the coupling of alkyne and allyl halide fragments, followed by a highly enantioselective epoxidation that sets the absolute configuration of the key chiral centers early in the sequence. This precision is maintained through a series of protective group manipulations using 2,2-dimethoxypropane, which shields sensitive hydroxyl groups during the critical oxidative cleavage of the terminal double bond via ozonolysis. The resulting aldehyde intermediate then undergoes a pivotal alpha-fluorination step mediated by a chiral pyrrolidine-based organocatalyst. This specific catalytic cycle ensures that the fluorine atom is introduced with high diastereoselectivity, preventing the formation of unwanted isomers that could compromise the biological activity of the final drug substance.
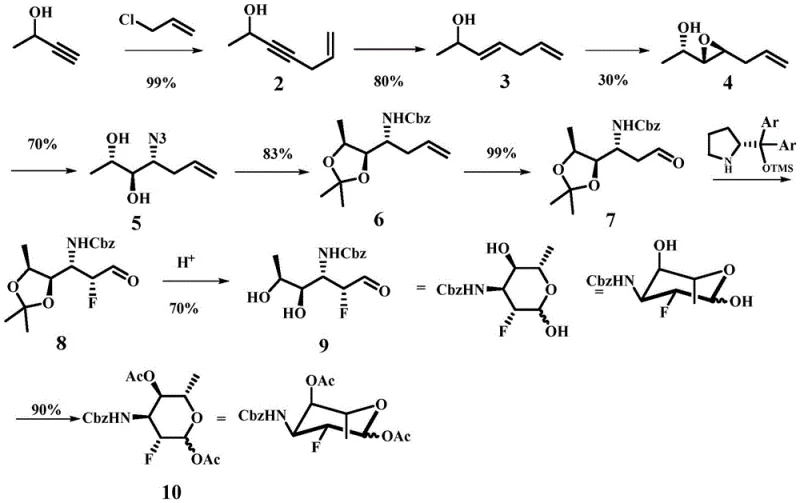
Furthermore, the impurity profile of the final product is rigorously controlled through the strategic design of the reaction pathway, which minimizes the generation of hard-to-remove byproducts. The use of specific acidic conditions for the final cyclization and deprotection steps ensures that the anomeric configuration is established correctly, yielding the desired alpha or beta anomers with high fidelity. For quality assurance teams, this mechanistic robustness translates to a cleaner crude product that requires less intensive chromatographic purification, thereby reducing solvent consumption and waste generation. The ability to tune the stereochemical outcome through the choice of chiral catalysts provides manufacturers with the flexibility to produce specific isomers required for different therapeutic applications, ensuring that the material meets the stringent purity specifications demanded by regulatory bodies for clinical use.
How to Synthesize 2-Fluoro-L-ristosamine Efficiently
The synthesis of this complex fluorinated sugar involves a multi-step sequence that transforms simple acyclic building blocks into a highly functionalized cyclic structure. The process initiates with a base-mediated coupling reaction to form the carbon backbone, followed by stereoselective reductions and epoxidations to install the necessary oxygen functionality. Subsequent steps involve the protection of diols, oxidative cleavage to generate an aldehyde, and the crucial organocatalytic fluorination that defines the molecule's unique properties. The final stages include acid-mediated deprotection and cyclization to close the pyranose ring, followed by acetylation to stabilize the anomeric center. Detailed standardized synthetic steps see the guide below.
- Perform alkaline coupling of 3-butyn-2-ol with 3-halopropene to form the initial alkyne intermediate.
- Execute Sharpless epoxidation followed by regioselective ring opening to establish stereochemistry.
- Conduct organocatalytic alpha-fluorination of the aldehyde intermediate followed by acid-mediated cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers substantial strategic benefits that extend beyond mere technical feasibility. The elimination of precious metal catalysts and toxic reagents directly correlates to a significant reduction in raw material costs and waste disposal expenses, enhancing the overall cost-efficiency of the manufacturing process. Moreover, the reliance on bulk commodity chemicals as starting materials mitigates the risk of supply shortages, ensuring a more resilient and reliable supply chain for downstream pharmaceutical production. The mild reaction conditions also imply lower energy consumption and reduced equipment wear, contributing to long-term operational savings and sustainability goals.
- Cost Reduction in Manufacturing: The process avoids the use of expensive transition metal catalysts and hazardous fluorinating agents like xenon difluoride, which are costly to procure and handle safely. By substituting these with organocatalysts and common reagents, the direct material costs are significantly lowered, while the simplified purification requirements reduce solvent usage and labor hours associated with chromatography. This structural cost advantage allows for more competitive pricing models in the supply of high-purity pharmaceutical intermediates without compromising on quality standards.
- Enhanced Supply Chain Reliability: Since the synthesis begins with widely available industrial chemicals such as 3-butyn-2-ol and allyl bromide, the dependency on niche or single-source suppliers is minimized. This diversification of the raw material base ensures that production schedules are less vulnerable to market fluctuations or geopolitical disruptions affecting specialized reagent availability. Consequently, manufacturers can maintain consistent lead times and fulfill large-volume orders with greater confidence, supporting the continuous operation of downstream drug formulation facilities.
- Scalability and Environmental Compliance: The synthetic route is designed with scalability in mind, utilizing unit operations that are easily transferable from laboratory to pilot and commercial scales. The absence of highly toxic byproducts and the use of greener catalytic systems align with increasingly strict environmental regulations, reducing the regulatory burden and permitting risks associated with chemical manufacturing. This compliance facilitates smoother audits and faster approval processes, enabling quicker time-to-market for new drug candidates relying on this critical sugar intermediate.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of 2-Fluoro-L-ristosamine. These answers are derived directly from the technical specifications and beneficial effects outlined in the patent documentation, providing clarity on the process capabilities and limitations. Understanding these details is essential for stakeholders evaluating the feasibility of integrating this intermediate into their existing drug development or manufacturing workflows.
Q: What are the key advantages of this synthesis method over traditional fluorination?
A: Unlike traditional methods requiring toxic xenon difluoride or harsh conditions, this patent utilizes mild organocatalytic fluorination and avoids precious metal catalysts, significantly reducing environmental impact and production costs while maintaining high stereoselectivity.
Q: Is this process scalable for industrial production of antibiotic intermediates?
A: Yes, the process relies on commercially available bulk raw materials like 3-butyn-2-ol and employs standard unit operations such as coupling, oxidation, and protection, making it highly suitable for commercial scale-up from kilogram to multi-ton quantities.
Q: How does this method ensure high purity for pharmaceutical applications?
A: The route incorporates Sharpless asymmetric epoxidation and chiral organocatalysis, which provide rigorous control over stereochemistry at multiple centers, ensuring the final 2-Fluoro-L-ristosamine meets stringent enantiomeric purity specifications required for API synthesis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Fluoro-L-ristosamine Supplier
At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that complex synthetic routes like the one described in CN109503681B are executed with precision and efficiency. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 2-Fluoro-L-ristosamine meets the highest international standards for pharmaceutical intermediates. We understand the critical nature of supply continuity in the drug development lifecycle and are committed to providing a stable, high-quality source of this valuable building block.
We invite you to contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific project requirements. By collaborating with us, you can gain access to specific COA data and comprehensive route feasibility assessments that will help optimize your supply chain strategy. Let us support your innovation with our manufacturing expertise and dedication to quality, ensuring your path from discovery to commercialization is smooth and successful.