Scalable Synthesis of Trifluoromethyl Indolopyranones for Antitumor Drug Development
The pharmaceutical industry is constantly seeking novel scaffolds that combine metabolic stability with potent biological activity, and patent CN114702504B presents a significant breakthrough in this domain through the development of trifluoromethyl substituted indolopyranone compounds. This technology addresses the critical need for efficient synthetic routes to complex heterocyclic systems that serve as core structures for next-generation antitumor agents. By fusing three distinct pharmacophores—indole, 2-pyrone, and the trifluoromethyl group—into a single molecular architecture, this invention offers a versatile platform for drug discovery. The strategic incorporation of the trifluoromethyl moiety is particularly noteworthy, as it enhances lipophilicity and modulates metabolic processes, which are essential parameters for optimizing drug candidates. As a reliable pharmaceutical intermediate supplier, understanding the structural nuances of these compounds is vital for partners aiming to develop robust pipelines in oncology.
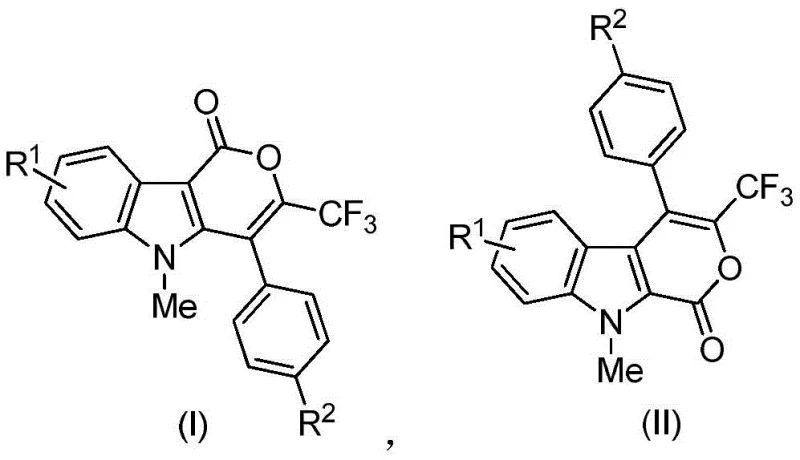
The scope of this technology covers a broad range of derivatives, defined by general formulas I and II, where substituents R1 and R2 can be varied to include alkyl, alkoxy, halogen, cyano, nitro, and ester groups. This structural diversity allows medicinal chemists to perform extensive structure-activity relationship (SAR) studies to pinpoint the most effective candidates for specific tumor types. The patent data highlights specific examples such as zyr-26, which demonstrated exceptional inhibitory activity against human glioblastoma cell lines. For R&D directors focused on purity and impurity profiles, the ability to access such a wide library of analogs through a unified synthetic strategy simplifies the lead optimization process significantly. Furthermore, the high regioselectivity reported in the patent ensures that the desired isomers are produced predominantly, reducing the burden on downstream purification efforts.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic strategies for constructing indolopyranone skeletons often rely on multi-step sequences that are both time-consuming and resource-intensive. These conventional methods frequently require the pre-activation of substrates, involving additional reagents and protection-deprotection steps that generate substantial stoichiometric waste. From a green chemistry perspective, these older routes suffer from poor atom economy, as a significant portion of the starting material mass ends up as byproducts rather than incorporated into the final drug molecule. Moreover, the harsh reaction conditions often associated with classical cyclization reactions can lead to the decomposition of sensitive functional groups, limiting the scope of substrates that can be utilized. For procurement managers, these inefficiencies translate directly into higher raw material costs and increased expenses for waste disposal, creating a bottleneck in the cost reduction in API manufacturing.
The Novel Approach
In stark contrast, the novel approach disclosed in the patent utilizes a transition metal-catalyzed direct C-H activation strategy that streamlines the synthesis into a single, efficient step. By employing a dichloro(pentamethylcyclopentadienyl)rhodium(III) dimer catalyst, the reaction achieves direct functionalization of inert carbon-hydrogen bonds without the need for prior substrate modification. This methodology not only simplifies the operational workflow but also drastically improves the overall yield, with many examples reporting excellent conversion rates under mild thermal conditions. The use of silver acetate as an oxidant facilitates the catalytic cycle effectively, ensuring high turnover numbers. This shift from multi-step classical synthesis to direct catalytic cyclization represents a paradigm shift in how complex heterocycles are manufactured, offering a pathway to substantial cost savings and enhanced supply chain reliability for high-purity pharmaceutical intermediates.
Mechanistic Insights into Rhodium-Catalyzed C-H Activation Cyclization
The core of this technological advancement lies in the sophisticated mechanism of Rhodium(III)-catalyzed C-H activation, which leverages the ortho-directing ability of the carboxyl group present on the N-methylindole starting material. The catalytic cycle initiates with the coordination of the rhodium species to the nitrogen or oxygen donor atoms, followed by the cleavage of the proximal C-H bond to form a stable metallacycle intermediate. This step is crucial as it determines the regioselectivity of the subsequent addition reaction. Once the metallacycle is formed, the trifluoromethyl substituted aryl alkyne inserts into the rhodium-carbon bond, setting the stage for the formation of the new carbon-carbon bonds that define the pyranone ring. The presence of the electron-withdrawing trifluoromethyl group on the alkyne influences the electronic density of the triple bond, facilitating this insertion step and ensuring high chemical selectivity.
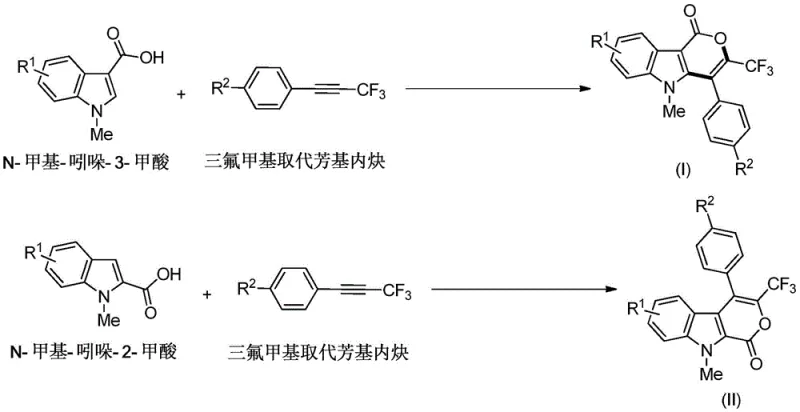
Following the alkyne insertion, the intermediate undergoes a reductive elimination or a similar cyclization step to close the lactone ring, releasing the final indolopyranone product and regenerating the active catalyst species in the presence of the oxidant. Understanding this mechanism is critical for controlling impurity profiles, as side reactions such as homocoupling of the alkyne or over-oxidation of the indole ring are minimized by the specific choice of catalyst and oxidant system. The patent emphasizes that the reaction conditions are mild enough to tolerate a wide variety of functional groups, including halogens and esters, which might be susceptible to degradation under more aggressive conditions. For technical teams, this mechanistic robustness means that scaling up the reaction from milligram to kilogram scale can be achieved with predictable outcomes, maintaining the high purity specifications required for clinical grade materials.
How to Synthesize Trifluoromethyl Indolopyranone Efficiently
The practical execution of this synthesis is designed to be straightforward, utilizing common laboratory equipment and readily available reagents to ensure ease of adoption in both research and production settings. The general procedure involves charging a reaction vessel with N-methylindole-3-carboxylic acid or its 2-carboxylic acid isomer, along with the appropriate trifluoromethyl substituted aryl alkyne. The catalyst system, consisting of the rhodium dimer and silver acetate, is then introduced into the mixture, followed by the addition of trifluoroethanol as the solvent of choice. The reaction mixture is sealed and heated to a temperature range of 70 to 100 degrees Celsius, typically requiring about 24 hours to reach full conversion as monitored by thin-layer chromatography. Detailed standardized synthesis steps are provided in the guide below to ensure reproducibility.
- Combine N-methylindole carboxylic acid, trifluoromethyl substituted aryl alkyne, Rhodium catalyst, and Silver acetate oxidant in trifluoroethanol solvent.
- Seal the reaction vessel and heat the mixture to 70-100°C with stirring for approximately 24 hours until TLC indicates completion.
- Remove solvent via rotary evaporation and purify the crude residue using silica gel column chromatography to isolate the pure indolopyranone product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement and supply chain leaders, the adoption of this synthetic route offers tangible benefits that extend beyond simple yield improvements, fundamentally altering the cost structure of producing these valuable intermediates. The elimination of pre-activation steps means fewer raw materials are required per unit of product, directly lowering the bill of materials. Furthermore, the reduction in synthetic steps decreases the cumulative loss of material that typically occurs during isolation and purification between stages, leading to a higher overall throughput from the same amount of starting feedstock. This efficiency gain is compounded by the use of a catalytic system that, while utilizing precious metals, operates with high turnover, minimizing the residual metal burden in the final product and reducing the cost associated with metal scavenging processes.
- Cost Reduction in Manufacturing: The streamlined nature of this one-pot cyclization reaction significantly reduces labor costs and energy consumption compared to traditional multi-step syntheses. By avoiding the use of hazardous reagents often required for substrate activation, the process also lowers the costs associated with safety compliance and specialized waste treatment. The high selectivity of the reaction minimizes the formation of difficult-to-remove byproducts, which simplifies the purification process and reduces the consumption of chromatography media and solvents. Consequently, the overall cost of goods sold (COGS) for these intermediates can be optimized, providing a competitive edge in the market for antitumor drug precursors.
- Enhanced Supply Chain Reliability: The starting materials for this reaction, specifically N-methylindole carboxylic acids and trifluoromethyl aryl alkynes, are commercially available from multiple global suppliers, ensuring a stable and resilient supply chain. This diversity in sourcing mitigates the risk of production delays caused by shortages of niche reagents. Additionally, the robustness of the reaction conditions allows for flexible manufacturing scheduling, as the process is not overly sensitive to minor fluctuations in temperature or pressure. This reliability is crucial for maintaining continuous production flows and meeting the strict delivery timelines demanded by pharmaceutical clients developing time-sensitive oncology therapies.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated to work efficiently with a wide range of substrates, which suggests that translation from pilot plant to commercial scale will encounter fewer technical hurdles. The use of trifluoroethanol as a solvent, while requiring recovery systems, is manageable within standard pharmaceutical manufacturing frameworks. Moreover, the significant reduction in stoichiometric waste aligns with increasingly stringent environmental regulations regarding chemical manufacturing. By adopting this greener synthetic route, companies can improve their sustainability metrics and reduce their environmental footprint, which is becoming a key factor in vendor selection for major multinational pharmaceutical corporations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these trifluoromethyl substituted indolopyranone compounds. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing clarity for potential partners evaluating this technology for their own development programs. Understanding these details is essential for making informed decisions about integrating these intermediates into your drug discovery pipeline.
Q: What are the key advantages of this Rhodium-catalyzed method over traditional synthesis?
A: This method eliminates the need for substrate pre-activation and utilizes direct C-H bond activation, resulting in superior atom economy, fewer synthetic steps, and significantly reduced stoichiometric waste compared to conventional multi-step strategies.
Q: What is the biological activity profile of these indolopyranone derivatives?
A: The synthesized compounds exhibit potent antitumor activity against various human cancer cell lines, including leukemia (HL60), glioma (U87), and breast cancer (MDA-MB-231), with specific derivatives showing IC50 values superior to cisplatin in certain assays.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process operates under mild conditions (70-100°C) using commercially available starting materials and standard purification techniques like column chromatography, making it highly adaptable for kilogram-to-ton scale production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Indolopyranone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the Rhodium-catalyzed C-H activation technology described in patent CN114702504B for the development of novel antitumor therapeutics. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless and efficient. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our capability to handle complex fluorinated chemistry and sensitive heterocyclic systems positions us as an ideal partner for bringing these promising candidates to the clinic.
We invite you to collaborate with us to leverage this advanced synthetic methodology for your specific project needs. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your volume requirements and timeline. We encourage you to reach out today to obtain specific COA data for our catalog of indolopyranone derivatives and to discuss route feasibility assessments for your custom synthesis projects. Together, we can accelerate the development of life-saving medicines by ensuring a reliable and cost-effective supply of these critical building blocks.