Revolutionizing Pyridine Synthesis: A Metal-Free One-Pot Strategy for High-Purity Intermediates
The pharmaceutical and fine chemical industries are constantly seeking more efficient, sustainable, and cost-effective pathways to construct complex heterocyclic scaffolds, particularly the ubiquitous pyridine ring which serves as a core structure in numerous bioactive molecules. A groundbreaking advancement in this domain is detailed in Chinese Patent CN111499565B, which discloses a novel preparation method for 2,3,4,6-tetra-substituted pyridine compounds. This technology represents a paradigm shift from traditional transition metal-catalyzed processes to a greener, metal-free multicomponent reaction strategy. By utilizing readily available starting materials such as diaryl ketones, aromatic aldehydes, and simple ketones in the presence of an amine source, this invention achieves high atom economy and operational simplicity. For R&D directors and procurement managers alike, this development signals a move towards cleaner synthetic routes that inherently avoid the regulatory and logistical burdens associated with heavy metal catalysts, positioning it as a critical innovation for reliable pharmaceutical intermediate supplier networks aiming for long-term sustainability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of highly substituted pyridine skeletons has relied heavily on sophisticated transition metal catalysis, which introduces significant complexities into the manufacturing workflow. Prior art methods often employ expensive and toxic catalysts such as Palladium (Pd), Rhodium (Rh), or Copper (Cu) to facilitate C-H functionalization and cyclization reactions. For instance, earlier methodologies described in literature utilize aminoalkenes and aryl iodides under Palladium catalysis, or N-sulfonyl ketimines and alkynes under Rhodium catalysis. While these methods can achieve decent yields, they suffer from severe drawbacks including the absolute necessity for rigorous removal of trace heavy metals to meet stringent pharmaceutical purity standards. Furthermore, the substrates required for these metal-catalyzed routes, such as specific alkynones or sulfonyl ketimines, are often not commercially available and require multi-step pre-synthesis, thereby inflating the overall cost of goods and extending the lead time for high-purity pharmaceutical intermediates. The reliance on inert atmospheres and specialized ligands further complicates the scale-up potential, making these conventional routes less attractive for cost reduction in API manufacturing.
The Novel Approach
In stark contrast to the metal-dependent legacy technologies, the method disclosed in CN111499565B offers a streamlined, one-pot solution that completely abolishes the use of metal catalysts. This innovative approach leverages a multicomponent reaction involving diaryl ketone compounds, aromatic aldehyde compounds, ketone compounds, and a simple amine source such as ammonium acetate. The reaction proceeds efficiently under air without the need for additional oxidants or inert gas protection, generating water as the only byproduct, which aligns perfectly with green chemistry principles. The substrate scope is remarkably broad, tolerating various functional groups including halogens, alkoxy groups, and heteroaryl moieties, ensuring wide applicability across different drug discovery programs. By eliminating the catalyst removal step and utilizing cheap, commodity-grade starting materials, this novel approach drastically simplifies the downstream processing and significantly enhances the economic viability of producing complex pyridine derivatives on a commercial scale.
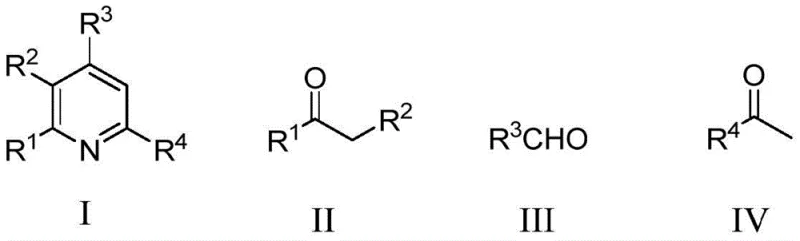
Mechanistic Insights into Metal-Free Multicomponent Cyclization
The mechanistic pathway of this transformation is a fascinating cascade of condensation and cyclization events that occur seamlessly in a single reaction vessel. As proposed in the patent, the reaction initiates with the condensation of the ketone compound (Formula IV) and the aromatic aldehyde (Formula III) to form a chalcone intermediate in situ. This chalcone then undergoes a Michael-type addition or condensation with the diaryl ketone (Formula II), setting the stage for ring closure. Subsequently, the amine source, typically ammonium acetate which releases ammonia under heating, participates in a cyclization event to form the dihydropyridine intermediate. The final aromatization to the stable pyridine ring is achieved through air oxidation, a process that is both thermodynamically favorable and operationally convenient. This intricate interplay of reactions highlights the elegance of the design, where the thermodynamic drive towards aromaticity powers the final step without requiring harsh chemical oxidants.
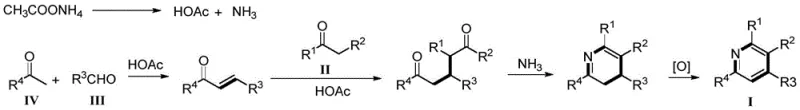
From an impurity control perspective, this metal-free mechanism offers distinct advantages for quality assurance teams. Since no transition metals are introduced, the risk of genotoxic impurities associated with metal leaching is entirely mitigated at the source. The primary impurities are likely to be unreacted starting materials or side products from the initial aldol condensation, which are generally easier to separate via standard crystallization or chromatography compared to metal complexes. The use of ammonium acetate as the nitrogen source ensures that the nitrogen incorporation is clean, avoiding the formation of complex amine byproducts often seen with primary amines. This inherent cleanliness of the reaction profile reduces the burden on analytical laboratories and simplifies the validation process for commercial scale-up of complex pharmaceutical intermediates, ensuring that the final product meets the rigorous specifications required for active pharmaceutical ingredient synthesis.
How to Synthesize 2,3,4,6-Tetrasubstituted Pyridines Efficiently
The practical execution of this synthesis is designed for robustness and ease of handling, making it accessible for both laboratory research and pilot plant operations. The process involves mixing the three carbonyl components—diaryl ketone, aromatic aldehyde, and ketone—with an excess of ammonium acetate in a polar aprotic solvent such as DMF or DMSO, or even under solvent-free conditions depending on the melting points of the reactants. The mixture is then heated to a moderate temperature range, typically between 80°C and 120°C, and stirred for a period of 15 to 25 hours. Upon completion, the reaction is quenched with water, and the product is extracted using common organic solvents like ethyl acetate. The crude material can be purified via silica gel column chromatography to afford the target pyridine derivative in high purity. For detailed standardized operating procedures and specific stoichiometric ratios tailored to your specific substrate, please refer to the comprehensive guide below.
- Mix diaryl ketone compounds, aromatic aldehyde compounds, ketone compounds, and an amine source such as ammonium acetate in a suitable reaction vessel.
- Heat the mixture to a temperature between 80°C and 120°C and stir for 15 to 25 hours to facilitate the multicomponent condensation and cyclization.
- Quench the reaction with water, extract the organic phase with ethyl acetate, dry over anhydrous sodium sulfate, and purify via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this metal-free technology translates into tangible strategic benefits that extend beyond simple chemistry. The most immediate impact is seen in the cost structure of the manufacturing process. By removing the requirement for precious metal catalysts like Palladium or Rhodium, which are subject to volatile market pricing and supply constraints, the raw material costs are significantly stabilized and reduced. Furthermore, the elimination of metal scavenging resins and the associated filtration equipment lowers the capital expenditure and operational overheads required for production. The use of commodity chemicals such as acetophenone and benzaldehyde ensures a secure and diversified supply base, reducing the risk of bottlenecks that often plague specialized reagent supply chains. This resilience is crucial for maintaining continuous production schedules and meeting the demanding delivery timelines of global pharmaceutical clients.
- Cost Reduction in Manufacturing: The economic model of this process is fundamentally superior due to the absence of expensive catalytic systems and the simplified workup procedure. Without the need for metal removal steps, the consumption of auxiliary materials such as scavengers and specialized filter aids is drastically curtailed, leading to substantial cost savings in the overall bill of materials. Additionally, the high atom economy of the multicomponent reaction means that a larger proportion of the input mass is converted into the desired product, minimizing waste disposal costs and maximizing yield efficiency. This lean manufacturing approach allows for a more competitive pricing strategy while maintaining healthy margins, which is essential in the highly price-sensitive generic pharmaceutical market.
- Enhanced Supply Chain Reliability: The reliance on widely available, bulk chemical feedstocks rather than bespoke, custom-synthesized building blocks greatly enhances supply chain security. Suppliers can source acetophenones and benzaldehydes from multiple vendors globally, mitigating the risk of single-source dependency. The robustness of the reaction conditions, which tolerate air and moisture, further reduces the logistical complexity of transporting and storing sensitive reagents. This flexibility ensures that production can be ramped up quickly in response to market demand surges without being hindered by long lead times for specialized catalysts or ligands, thereby guaranteeing a steady flow of high-quality intermediates to downstream customers.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this process is exceptionally well-suited for large-scale implementation. The generation of water as the primary byproduct and the avoidance of toxic heavy metals simplify wastewater treatment and reduce the environmental footprint of the facility. Regulatory compliance becomes more straightforward as the burden of proving low residual metal levels in the final API is removed. The mild reaction temperatures and atmospheric pressure conditions also lower the energy consumption and safety risks associated with high-pressure hydrogenation or cryogenic reactions, facilitating a smoother technology transfer from the lab to the kiloton scale.
Frequently Asked Questions (FAQ)
To address common inquiries regarding the technical feasibility and commercial implementation of this synthesis method, we have compiled a set of answers based on the detailed experimental data provided in the patent documentation. These insights cover critical aspects such as substrate compatibility, reaction optimization, and purification strategies, offering a clear picture of what partners can expect when integrating this technology into their supply chain. Understanding these nuances is vital for making informed decisions about process adoption and resource allocation.
Q: What are the primary advantages of this metal-free pyridine synthesis method compared to traditional transition metal catalysis?
A: The primary advantage is the complete elimination of heavy metal catalysts like Palladium or Rhodium, which removes the risk of toxic metal residues in the final pharmaceutical product and eliminates the costly purification steps required to remove these metals.
Q: What is the typical yield range for this one-pot synthesis of tetrasubstituted pyridines?
A: According to the patent data, the method demonstrates robust yields, typically ranging from 60% to 89% depending on the specific substituents, with many examples achieving yields above 80% under optimized conditions.
Q: Is this process scalable for industrial production of API intermediates?
A: Yes, the process uses cheap and readily available raw materials like acetophenone and benzaldehyde, operates under air without inert gas protection, and involves simple workup procedures, making it highly suitable for large-scale commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2,3,4,6-Tetrasubstituted Pyridines Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this metal-free synthesis route for the next generation of pharmaceutical intermediates. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to industrial manufacturing is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 2,3,4,6-tetrasubstituted pyridines meets the highest international standards. We are committed to leveraging this innovative chemistry to deliver cost-effective and high-quality solutions that accelerate our clients' drug development timelines.
We invite you to explore how this advanced synthetic methodology can optimize your current supply chain and reduce your overall manufacturing costs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate the viability of this approach for your target molecules, ensuring a partnership built on transparency, quality, and mutual success.