Advanced Synthesis of Brivaracetam Acid Impurity for Global Pharmaceutical Quality Control
The pharmaceutical industry faces relentless pressure to ensure the safety and efficacy of active pharmaceutical ingredients (APIs), particularly for potent antiepileptic drugs like Brivaracetam. As regulatory bodies such as the FDA and EMA tighten guidelines on impurity profiling, the ability to synthesize specific degradation products and process impurities in high purity becomes a critical bottleneck in drug development. Patent CN114213306A addresses this challenge by disclosing a robust preparation method for Brivaracetam acid impurity, specifically (S)-2-[(R)-2-oxo-4-propylpyrrolidin-1-yl]butyric acid. This technical breakthrough offers a streamlined alternative to traditional isolation methods, which often suffer from low yields and poor stereochemical control. By establishing a dedicated synthetic route rather than relying on trace isolation from reaction mixtures, manufacturers can secure a reliable supply of reference standards essential for validating analytical methods and ensuring batch-to-batch consistency in commercial production.
For R&D directors and quality control managers, the significance of this patent lies in its ability to generate sufficient quantities of the target impurity without compromising optical purity. In the context of Brivaracetam, a third-generation antiepileptic agent, even trace levels of hydrolysis impurities can impact patient safety profiles. The traditional approach of attempting to isolate this impurity from the final drug substance synthesis is fraught with difficulties, including the inability to control alkali dosage and the propensity for racemization under strongly acidic or basic conditions. The methodology outlined in CN114213306A circumvents these pitfalls by constructing the molecule through a controlled two-step sequence that prioritizes chiral retention and operational simplicity, thereby setting a new benchmark for impurity management in neurology drug manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the acquisition of Brivaracetam acid impurity has been a significant pain point for pharmaceutical developers, primarily due to the reliance on inefficient isolation techniques or non-selective hydrolysis of the parent drug. Conventional literature methods often involve subjecting Brivaracetam to harsh hydrolysis conditions, which inevitably leads to the formation of the acid impurity in only trace amounts alongside a complex matrix of byproducts. This makes the subsequent purification process exceedingly difficult, often requiring preparative HPLC or chiral chromatography to separate the desired impurity from the unreacted starting material and other degradation products. Furthermore, the use of solvents like dichloromethane in previous synthetic attempts has been linked to increased rates of racemization, jeopardizing the stereochemical integrity required for accurate toxicological assessment. These limitations result in prohibitively high costs and extended lead times, creating a supply chain vulnerability for companies needing gram-to-kilogram quantities of the impurity for stability studies and regulatory filings.
The Novel Approach
In stark contrast to these legacy methods, the invention described in CN114213306A introduces a constructive synthetic strategy that builds the impurity molecule from advanced intermediates rather than degrading the final API. This novel approach utilizes a condensation reaction between a specific acyl halide derivative and an optically pure amino acid ester, followed by a mild alkaline hydrolysis. By selecting acetonitrile as the reaction solvent and incorporating phase transfer catalysts, the process effectively suppresses racemization, a common failure mode in peptide-like bond formations. The result is a high-yielding pathway that delivers the target impurity with purity levels exceeding 99%, completely bypassing the need for chiral column resolution. This shift from isolation to dedicated synthesis represents a paradigm change in how critical quality attributes are managed, offering a scalable solution that aligns perfectly with the rigorous demands of modern GMP manufacturing environments.
Mechanistic Insights into Condensation and Hydrolysis Strategy
The core of this technological advancement rests on a meticulously optimized two-step reaction mechanism designed to preserve chirality while maximizing throughput. The first step involves the nucleophilic acyl substitution where the acyl chloride (Formula 2) reacts with (S)-2-aminobutyric acid ester hydrochloride (Formula 3). This reaction is conducted in the presence of a strong base, such as potassium hydroxide, and a phase transfer catalyst like polyethylene glycol (PEG400). The role of the phase transfer catalyst is pivotal; it facilitates the transport of the anionic nucleophile into the organic phase, enhancing the reaction rate without necessitating extreme temperatures that could induce epimerization. The use of molecular sieves as a water scavenger further drives the equilibrium towards product formation by removing the water generated during the neutralization of the amine salt, ensuring high conversion rates of the limiting reagent.
Following the formation of the intermediate ester (Formula 4), the second step employs a controlled hydrolysis to reveal the free carboxylic acid. Unlike the harsh conditions often used in degradation studies, this process utilizes lithium hydroxide or sodium hydroxide in a methanol-water mixture at moderate temperatures ranging from 0°C to 50°C. This mild alkaline environment is sufficient to cleave the ester bond but gentle enough to prevent the opening of the pyrrolidone ring or the inversion of the chiral centers at the alpha-position of the butyric acid moiety. The preservation of the (S) configuration at the side chain and the (R) configuration on the pyrrolidone ring is critical, as any deviation would render the reference standard useless for chiral HPLC method validation. This mechanistic precision ensures that the final product is chemically and stereochemically identical to the process-related impurity found in Brivaracetam synthesis.
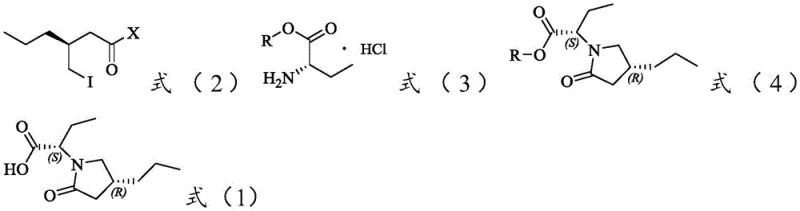
The strategic selection of reagents and conditions in this patent directly addresses the impurity control mechanisms required for regulatory compliance. By avoiding the use of dichloromethane, which was identified in the background art as a promoter of racemization, the inventors have secured a more robust process window. The workup procedure, involving acid washing to remove excess amine and base, followed by crystallization from ethyl acetate and isopropyl ether, provides an additional layer of purification. This crystallization step is not merely a isolation technique but a critical purification unit operation that rejects structurally related byproducts and residual solvents. For R&D teams, understanding this mechanism allows for better prediction of potential genotoxic impurities or residual solvent issues, facilitating a smoother path through the Investigational New Drug (IND) application process.
How to Synthesize Brivaracetam Acid Impurity Efficiently
The synthesis protocol detailed in the patent provides a clear roadmap for laboratory and pilot-scale production, emphasizing reproducibility and safety. The process begins with the activation of the base and drying agents in acetonitrile, followed by the sequential addition of the chiral amine salt and the acylating agent under strict temperature control. This order of addition is crucial to minimize the exposure of the reactive acyl chloride to moisture and to control the exotherm associated with amide bond formation. After the condensation is complete, the intermediate is isolated via crystallization, which serves as a hold point for quality control before proceeding to the final hydrolysis. The hydrolysis step is equally straightforward, utilizing common reagents like lithium hydroxide monohydrate, making the entire sequence accessible to standard multipurpose chemical manufacturing facilities without the need for specialized equipment.
- Perform a condensation reaction between the acyl halide compound (Formula 2) and (S)-2-aminobutyric acid ester hydrochloride (Formula 3) in acetonitrile with a base and phase transfer catalyst to form the intermediate ester (Formula 4).
- Hydrolyze the intermediate ester (Formula 4) under alkaline conditions using lithium hydroxide or sodium hydroxide in a methanol/water mixture.
- Adjust the pH to neutral, extract with organic solvent, and crystallize the final product (Formula 1) to achieve high purity without chiral resolution.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers profound advantages for procurement managers and supply chain heads tasked with optimizing the cost of goods sold (COGS) for API production. The primary value driver is the elimination of chiral chromatography, a notoriously expensive and low-throughput purification technique. By achieving high optical purity through synthetic control rather than downstream separation, manufacturers can drastically reduce the consumption of chiral stationary phases and the associated solvent volumes. This translates into significant cost savings in both material procurement and waste disposal, directly impacting the bottom line. Furthermore, the use of commodity chemicals such as acetonitrile, potassium hydroxide, and methanol ensures that the raw material supply chain is resilient and less susceptible to market volatility compared to processes relying on exotic catalysts or reagents.
- Cost Reduction in Manufacturing: The economic benefits of this process are substantial, primarily driven by the simplification of the purification train. Traditional methods that rely on isolating impurities from reaction mixtures often require multiple chromatographic steps, which are labor-intensive and consume vast amounts of solvents. In contrast, this novel method relies on crystallization as the primary purification tool, a unit operation that is inherently cheaper and easier to scale. The removal of the chiral column requirement alone represents a massive reduction in operational expenditure, as chiral resins are costly and have limited lifecycles. Additionally, the high yield of the intermediate step means that less starting material is wasted, improving the overall atom economy of the process and reducing the cost per gram of the final reference standard.
- Enhanced Supply Chain Reliability: Supply chain continuity is paramount for pharmaceutical companies facing tight regulatory deadlines. This synthesis route enhances reliability by utilizing readily available starting materials that are not subject to complex geopolitical supply constraints. The reaction conditions are mild and do not require cryogenic temperatures below -20°C or high-pressure equipment, meaning the process can be executed in a wide range of standard chemical reactors. This flexibility allows for easier technology transfer between different manufacturing sites and reduces the risk of production delays caused by equipment availability. Moreover, the robustness of the reaction against racemization reduces the likelihood of batch failures due to out-of-specification optical purity, ensuring a consistent and predictable supply of the critical impurity standard.
- Scalability and Environmental Compliance: As the demand for Brivaracetam grows globally, the ability to scale impurity synthesis to meet commercial needs is essential. This process is designed with scalability in mind, using solvents and reagents that are compatible with large-scale industrial operations. The replacement of dichloromethane with acetonitrile also aligns with modern environmental, health, and safety (EHS) guidelines, as acetonitrile is generally preferred in green chemistry metrics over chlorinated solvents. This shift reduces the environmental footprint of the manufacturing process and simplifies waste stream management, lowering the costs associated with hazardous waste disposal. The straightforward workup and crystallization steps further facilitate scale-up, allowing manufacturers to increase batch sizes from kilograms to tons without encountering the mixing or heat transfer issues often seen in chromatographic separations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and claims presented in patent CN114213306A, providing clarity for technical teams evaluating the feasibility of adopting this method for their quality control laboratories or pilot plants. Understanding these nuances is key to leveraging the full potential of the technology for impurity management.
Q: Why is acetonitrile preferred over dichloromethane for this synthesis?
A: According to patent CN114213306A, using acetonitrile significantly reduces the risk of racemization compared to dichloromethane, ensuring the stereochemical integrity of the chiral centers in the final impurity standard.
Q: Does this method require chiral column chromatography?
A: No, the disclosed method utilizes chiral starting materials and mild reaction conditions that preserve optical purity, eliminating the need for expensive and time-consuming chiral column resolution.
Q: What are the typical reaction temperatures for the condensation step?
A: The condensation reaction is optimally performed at low temperatures, specifically between -10°C and 10°C, to control exothermicity and minimize side reactions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Brivaracetam Acid Impurity Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your drug substance depends on the quality of your reference standards and impurities. Our team of expert chemists has extensively analyzed the route disclosed in CN114213306A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this chemistry to life. We are equipped with stringent purity specifications and rigorous QC labs capable of verifying the stereochemical purity of complex molecules like Brivaracetam acid impurity using state-of-the-art chiral HPLC and NMR spectroscopy. Our commitment to quality ensures that every batch we deliver meets the exacting standards required for global regulatory submissions, providing you with the confidence needed to advance your drug development programs.
We invite you to collaborate with us to optimize your supply chain for Brivaracetam and related neurological therapeutics. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis that evaluates how switching to this synthetic route can reduce your overall impurity sourcing costs. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project's unique requirements. Let us handle the complexities of synthesis and purification so you can focus on delivering life-saving medications to patients worldwide.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →