Scalable Metal-Free Synthesis of Thiazolidin-4-one Derivatives for Pharmaceutical Applications
Introduction to Advanced Metal-Free Heterocycle Synthesis
The landscape of pharmaceutical intermediate manufacturing is constantly evolving towards greener, more cost-effective methodologies that do not compromise on purity or yield. A significant breakthrough in this domain is documented in patent CN110054597B, which discloses a novel preparation method for thiazolidin-4-one derivatives synthesized under strictly metal-free conditions. This technology addresses critical pain points in the production of bioactive scaffolds known for their antibacterial, antiviral, and anticancer properties. By leveraging a unique cyclization strategy that bypasses the need for traditional transition metal catalysts, this innovation offers a streamlined pathway to high-value heterocyclic compounds. The methodology utilizes simple, commercially available starting materials and operates under mild thermal conditions, representing a substantial leap forward in process chemistry efficiency. For R&D teams and procurement strategists alike, understanding the implications of this patent is vital for optimizing supply chains and reducing the overall cost of goods sold in drug development pipelines.
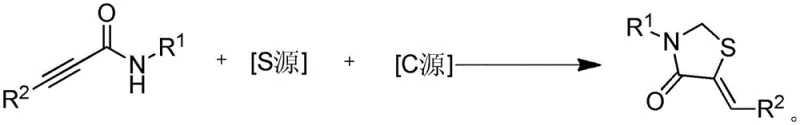
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of thiazolidin-4-one scaffolds has relied heavily on multi-step procedures or catalytic systems that introduce significant economic and environmental burdens. Traditional approaches often involve the condensation of Schiff bases with mercaptoacetic acid, a route that is notoriously cumbersome and generates substantial waste due to the multiple isolation steps required. Furthermore, more modern attempts to streamline this synthesis have frequently depended on the use of transition metal catalysts, specifically copper or noble metals like palladium, to facilitate the cyclization process. While these metal-catalyzed methods can improve reaction rates, they introduce severe downstream challenges, including the necessity for rigorous heavy metal removal to meet stringent pharmaceutical purity standards. The cost of these precious metal catalysts, coupled with the expense of specialized ligands and scavengers, drastically inflates the production budget. Additionally, the presence of residual metals poses a regulatory risk, often requiring extensive analytical validation and additional purification cycles that extend lead times and reduce overall throughput.
The Novel Approach
In stark contrast to these legacy methods, the technology outlined in patent CN110054597B presents a direct, one-pot synthesis route that completely eliminates the need for any metal catalyst. This novel approach utilizes N-aryl(alkyl)-3-arylpropynamides as the core substrate, reacting them with inexpensive inorganic sulfur sources such as sodium sulfide or potassium sulfide. Crucially, the reaction employs common halogenated solvents like dichloromethane or dibromomethane not merely as a medium, but actively as the carbon source for the ring construction. This dual functionality of the solvent simplifies the reagent list and enhances atom economy. The reaction proceeds smoothly at a moderate temperature of 80°C under atmospheric pressure, avoiding the need for specialized high-pressure reactors or cryogenic conditions. By removing the dependency on precious metals, this method inherently produces a cleaner crude product profile, significantly reducing the complexity of the workup procedure and enabling a more sustainable manufacturing process that aligns with modern green chemistry principles.
Mechanistic Insights into Metal-Free Cyclization
The mechanistic pathway of this transformation is both elegant and efficient, relying on fundamental organic reactivity rather than complex metal-ligand interactions. The reaction initiates with the nucleophilic attack of the sulfide anion on the electron-deficient alkyne moiety of the propynamide substrate. This electrophilic addition generates a reactive vinyl sulfide intermediate, which sets the stage for the subsequent ring closure. Unlike metal-catalyzed processes that often require oxidative addition and reductive elimination cycles, this metal-free route proceeds through a concerted series of ionic transformations. The intermediate undergoes a crucial intramolecular [1,4]-hydrogen migration, activating the nitrogen center for the final cyclization step. This migration is facilitated by the specific electronic environment created by the carbonyl group and the adjacent sulfur atom, ensuring high regioselectivity for the desired thiazolidin-4-one core.
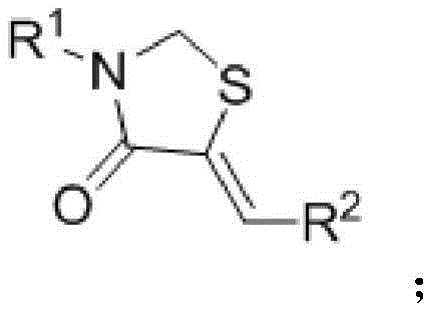
Following the hydrogen migration, the activated nitrogen species engages in a nucleophilic substitution reaction with the dihalomethane carbon source. This step effectively closes the five-membered ring, incorporating the methylene bridge essential for the thiazolidine structure. The absence of metal catalysts means that the reaction mixture is free from transition metal contaminants, which is a massive advantage for impurity control. In traditional catalysis, metal residues can catalyze unwanted side reactions or form stable complexes with the product, leading to difficult-to-remove impurities. Here, the impurity profile is dominated primarily by unreacted starting materials or simple hydrolysis byproducts, which are far easier to separate via standard crystallization or chromatography. This clean reaction profile ensures that the final high-purity pharmaceutical intermediates meet the rigorous specifications required for downstream drug substance manufacturing without the need for expensive metal scavenging resins.
How to Synthesize Thiazolidin-4-one Derivatives Efficiently
Implementing this synthesis route in a laboratory or pilot plant setting requires careful attention to stoichiometry and solvent quality, although the operational parameters are remarkably forgiving. The process begins by dissolving the N-arylpropynamide substrate in a polar aprotic solvent such as tetrahydrofuran, acetonitrile, or 1,4-dioxane. To this solution, the sulfur source, typically sodium sulfide, is added in a slight molar excess to ensure complete conversion of the alkyne. The carbon source, dichloromethane, is then introduced, serving both as a reactant and a co-solvent. The detailed standardized synthesis steps, including specific mixing orders and quenching protocols, are provided in the guide below to ensure reproducibility and safety during scale-up operations.
- Mix N-aryl(alkyl)-3-arylpropynamide substrate with sodium sulfide or potassium sulfide as the sulfur source in a suitable solvent like THF.
- Add dichloromethane or dibromomethane as the carbon source and heat the reaction mixture to 80°C under atmospheric pressure.
- Maintain stirring for 14-16 hours, then filter, concentrate, and purify via silica gel column chromatography to isolate the target derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this metal-free synthesis technology translates directly into tangible bottom-line improvements and risk mitigation. The most immediate impact is seen in the raw material cost structure, where the elimination of palladium and copper catalysts removes a volatile and expensive line item from the bill of materials. Furthermore, the reliance on commodity chemicals like sodium sulfide and dichloromethane ensures a stable and resilient supply chain, as these reagents are produced globally in massive quantities and are not subject to the geopolitical supply constraints often associated with rare earth metals or specialized ligands. This stability allows for more accurate long-term forecasting and inventory planning, reducing the risk of production stoppages due to material shortages.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven primarily by the drastic simplification of the downstream processing workflow. In conventional metal-catalyzed routes, a significant portion of the manufacturing cost is attributed to the purification steps required to reduce metal residues to parts-per-million levels, often involving costly scavenger resins and multiple filtration cycles. By operating under metal-free conditions, this patent enables a streamlined workup where the product can often be isolated through simple filtration and solvent evaporation followed by basic recrystallization. This reduction in unit operations lowers energy consumption, reduces solvent usage, and minimizes labor hours, resulting in substantial cost savings per kilogram of produced intermediate. Additionally, the high atom economy of the reaction ensures that a greater proportion of the input mass is converted into valuable product rather than waste, further enhancing the overall process efficiency.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route significantly de-risks the supply chain for critical pharmaceutical intermediates. Because the reaction does not depend on sensitive catalysts that may degrade upon exposure to air or moisture, the reagents have longer shelf lives and are easier to store and transport. This tolerance to ambient conditions reduces the need for specialized storage infrastructure and allows for more flexible logistics arrangements. Moreover, the broad substrate scope demonstrated in the patent means that a single manufacturing platform can be adapted to produce a wide variety of derivatives by simply swapping the starting propynamide. This versatility allows manufacturers to respond quickly to changing market demands or clinical trial requirements without needing to requalify entirely new synthetic processes, thereby ensuring continuous supply continuity for diverse drug development programs.
- Scalability and Environmental Compliance: From an environmental and regulatory perspective, this technology offers a clear path to sustainable commercial scale-up of complex heterocycles. The reaction operates at atmospheric pressure and moderate temperatures, eliminating the safety hazards associated with high-pressure hydrogenation or exothermic metal-catalyzed couplings. This inherent safety profile simplifies the engineering controls required for large-scale reactors, lowering capital expenditure for new production lines. Furthermore, the absence of heavy metals drastically reduces the toxicity of the waste stream, simplifying wastewater treatment and disposal compliance. The process generates minimal hazardous waste compared to traditional multi-step syntheses, aligning with increasingly strict global environmental regulations and corporate sustainability goals. This eco-friendly profile not only reduces disposal costs but also enhances the brand reputation of the manufacturer as a responsible partner in the pharmaceutical value chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this metal-free synthesis technology. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the practical application of this method for industrial partners seeking to optimize their production capabilities.
Q: What are the primary advantages of this metal-free synthesis route?
A: The primary advantage is the complete elimination of expensive transition metal catalysts like copper or palladium, which significantly reduces raw material costs and simplifies downstream purification by removing the need for heavy metal scavenging steps.
Q: What represents the key starting materials for this reaction?
A: The reaction utilizes readily available N-aryl(alkyl)-3-arylpropynamides as substrates, combined with inexpensive inorganic sulfur sources such as sodium sulfide and common halogenated solvents like dichloromethane acting as the carbon source.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process operates under mild conditions at atmospheric pressure and moderate temperatures around 80°C, utilizing common solvents and avoiding hazardous high-pressure equipment, making it highly amenable to commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Thiazolidin-4-one Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of metal-free synthesis technologies in modernizing the production of critical drug scaffolds. As a premier CDMO partner, we possess the technical expertise to rapidly adapt and optimize the pathway described in patent CN110054597B for your specific project needs. Our facilities are equipped to handle the commercial scale-up of complex pharmaceutical intermediates, with extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We maintain stringent purity specifications and operate rigorous QC labs to ensure that every batch of thiazolidin-4-one derivative meets the highest international standards for potency and impurity profiles, giving you confidence in the quality of your supply chain.
We invite you to collaborate with us to leverage this cost-effective and environmentally friendly synthesis route for your next development program. Our team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this metal-free approach can improve your margins. Please contact our technical procurement team today to request specific COA data for our existing library of heterocycles or to discuss route feasibility assessments for your proprietary targets, and let us help you accelerate your path to market with superior chemistry.