Advanced One-Step Trifluoromethylation for High-Purity 3-Chloro-6-Trifluoromethylpyridazine Manufacturing
The pharmaceutical and agrochemical industries are constantly seeking more efficient pathways to introduce trifluoromethyl groups into heterocyclic scaffolds, a modification known to drastically enhance metabolic stability and lipophilicity. Patent CN102532036A presents a groundbreaking methodology for the preparation of 3-chloro-6-trifluoromethylpyridazine, a critical building block for various bioactive compounds. This innovation addresses the long-standing challenges associated with trifluoromethylation by replacing complex multi-step sequences with a streamlined, high-yield one-step reaction. By leveraging a specific palladium and copper dual-catalyst system, the process achieves superior conversion rates while utilizing commercially accessible reagents. For R&D directors and procurement specialists, this patent represents a significant opportunity to optimize supply chains for high-purity pharmaceutical intermediates, ensuring both cost efficiency and reliable availability for downstream drug synthesis.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the introduction of a trifluoromethyl group onto a pyridazine ring has been a cumbersome and economically inefficient process. Conventional synthetic routes typically rely on a two-step sequence where 3-chloro-6-iodopyridazine is first converted into a boronic acid derivative via Suzuki coupling. This intermediate must then undergo an oxidative coupling reaction with trifluoromethylating agents such as CF3SiMe3 in the presence of copper catalysts. The critical drawback of this traditional approach lies in its stringent requirement for absolutely anhydrous conditions during the second step, which necessitates specialized equipment and rigorous operational controls. Furthermore, the reagents involved, particularly copper triflate complexes and silyl-based trifluoromethyl sources, are notoriously expensive and difficult to source in bulk quantities. Consequently, these legacy methods often suffer from mediocre overall yields, typically hovering around 60%, which generates significant waste and inflates the cost of goods sold for the final active pharmaceutical ingredient.
The Novel Approach
In stark contrast to the cumbersome legacy protocols, the method disclosed in CN102532036A revolutionizes the synthesis by condensing the entire transformation into a single, robust reaction step. This novel approach utilizes methyl 2,2-difluoro-2-(fluorosulfonyl)acetate as the trifluoromethyl source, reacting directly with 3-chloro-6-iodopyridazine under relatively mild thermal conditions. The elimination of the intermediate boronic acid formation step not only reduces the total processing time but also removes the need for sensitive anhydrous environments, thereby simplifying the engineering requirements for production reactors. By achieving yields in the range of 80% to 90%, this method significantly maximizes atom economy and minimizes the generation of chemical waste. The use of diluted hydrofluoric acid as a solvent further enhances the practicality of the process, offering a balance between reactivity and safety that is conducive to large-scale manufacturing operations.
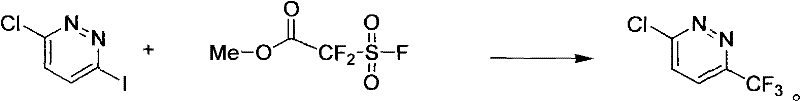
Mechanistic Insights into Pd/Cu Catalyzed Trifluoromethylation
The core of this technological breakthrough lies in the sophisticated interplay between the palladium and copper catalysts within the reaction matrix. The [1,1'-bis(diphenylphosphino)ferrocene]palladium(II) dichloride complex acts as the primary driver for the oxidative addition of the aryl iodide bond, activating the pyridazine ring for subsequent functionalization. Simultaneously, the cupric iodide co-catalyst facilitates the transfer of the trifluoromethyl group from the fluorosulfonyl precursor to the activated palladium center. This dual-catalytic cycle ensures that the reaction proceeds with high regioselectivity, preventing the formation of unwanted isomers or dehalogenated byproducts that often plague trifluoromethylation reactions. The stability of the ferrocene-based ligand system allows the catalyst to maintain activity over extended reaction times of 20 to 36 hours, ensuring complete conversion of the starting material even at the substantial scale required for commercial production.
Beyond the primary catalytic cycle, the mechanism inherently supports superior impurity control, a critical factor for R&D directors focused on regulatory compliance. The specific choice of reagents and the single-step nature of the reaction minimize the generation of complex side products that are difficult to separate during purification. Unlike the multi-step Suzuki route, which can accumulate boron-containing impurities and silyl residues, this direct trifluoromethylation pathway results in a cleaner crude reaction mixture. The subsequent workup involving ethyl acetate extraction and recrystallization is highly effective at removing residual catalyst metals and unreacted starting materials. This inherent purity profile reduces the burden on downstream purification processes, ensuring that the final 3-chloro-6-trifluoromethylpyridazine meets the stringent quality specifications required for use in sensitive pharmaceutical applications without extensive additional processing.
How to Synthesize 3-Chloro-6-Trifluoromethylpyridazine Efficiently
Implementing this synthesis route in a production environment requires careful attention to the stoichiometry of the catalyst system and the thermal profile of the reaction. The patent outlines a procedure where the reactants are combined under a nitrogen atmosphere to prevent oxidative degradation of the catalysts, followed by heating to temperatures between 90°C and 110°C. The detailed standardized synthesis steps, including precise molar ratios and specific workup parameters, are critical for reproducing the high yields reported in the patent embodiments. For process chemists looking to adopt this technology, adhering to the specified reaction times of 20 to 36 hours is essential to ensure full conversion while maintaining the integrity of the product. The following guide provides the structural framework for executing this synthesis, ensuring that the transition from laboratory scale to commercial manufacturing is seamless and reproducible.
- Charge the reactor with 3-chloro-6-iodopyridazine and methyl 2,2-difluoro-2-(fluorosulfonyl)acetate under nitrogen protection.
- Add the dual catalyst system comprising cupric iodide and [1,1'-bis(diphenylphosphino)ferrocene]palladium(II) dichloride complex in diluted hydrofluoric acid solvent.
- Heat the mixture to 90-110°C for 20-36 hours, then perform ethyl acetate extraction and recrystallization to isolate the high-purity product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology offers profound strategic advantages that extend far beyond simple chemical efficiency. The shift from a two-step, anhydrous-sensitive process to a one-step, robust reaction fundamentally alters the cost structure and risk profile of manufacturing this key intermediate. By eliminating the need for expensive and hard-to-source reagents like copper triflate and trimethylsilyl trifluoromethane, the raw material costs are drastically reduced. Furthermore, the simplification of the process flow means that production capacity can be increased without significant capital investment in new specialized equipment, as standard reactors capable of handling mild heating and standard solvents are sufficient. This operational flexibility allows for more responsive supply chain management, enabling manufacturers to scale production up or down based on market demand without incurring prohibitive changeover costs or delays.
- Cost Reduction in Manufacturing: The economic benefits of this new route are driven primarily by the consolidation of two reaction steps into one, which inherently cuts labor, energy, and solvent consumption by nearly half. The replacement of precious and specialized catalysts with commercially available cupric iodide and palladium complexes further drives down the direct material costs, making the final product more price-competitive in the global market. Additionally, the higher yield of 80-90% compared to the traditional 60% means that less raw material is wasted per kilogram of product, significantly improving the overall material efficiency and reducing the cost of waste disposal. These cumulative factors result in a substantially lower cost of goods sold, providing a strong margin buffer for downstream pharmaceutical manufacturers.
- Enhanced Supply Chain Reliability: Supply chain resilience is significantly bolstered by the use of reagents that are readily available from multiple global chemical suppliers, reducing the risk of single-source bottlenecks. The removal of strict anhydrous requirements simplifies logistics and storage, as the reagents do not require specialized moisture-free containment during transport or handling. This robustness ensures that production schedules are less likely to be disrupted by minor environmental fluctuations or supply delays of niche reagents. Consequently, suppliers can offer more reliable lead times and consistent delivery schedules, which is critical for pharmaceutical companies managing tight production timelines for active drug substances.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, the reduction in synthetic steps directly correlates to a smaller environmental footprint, aligning with modern green chemistry principles. The process generates less chemical waste and utilizes solvents that are easier to recover and recycle, simplifying compliance with increasingly stringent environmental regulations. The thermal stability of the reaction allows for safe scale-up from kilogram to multi-ton batches without the exothermic risks often associated with complex coupling reactions. This scalability ensures that the supply of high-purity pharmaceutical intermediates can grow in tandem with the commercial success of the final drug product, securing long-term supply continuity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this trifluoromethylation technology. These answers are derived directly from the technical specifications and beneficial effects outlined in the patent documentation, providing clarity on how this method compares to industry standards. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios. The insights provided here aim to bridge the gap between theoretical patent claims and practical industrial application, ensuring that all stakeholders have a clear understanding of the process capabilities.
Q: How does this new trifluoromethylation method improve upon traditional Suzuki coupling routes?
A: Traditional methods require a two-step process involving boronic acid formation followed by oxidative coupling under strictly anhydrous conditions, often yielding only 60%. This patent introduces a direct one-step reaction that eliminates the need for expensive silyl reagents and complex anhydrous setups, significantly boosting yield to the 80-90% range while simplifying the operational workflow.
Q: What specific catalyst system is utilized to achieve high conversion rates?
A: The process employs a robust dual-catalyst system consisting of cupric iodide (CuI) and [1,1'-bis(diphenylphosphino)ferrocene]palladium(II) dichloride complex. These catalysts are commercially available and cost-effective compared to the specialized copper triflate complexes required in prior art, facilitating easier procurement and lower production costs.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the method is highly scalable due to its single-step nature and the use of readily available solvents like diluted hydrofluoric acid. The elimination of sensitive anhydrous requirements and the high thermal stability of the reaction conditions (90-110°C) make it ideal for kilogram-to-ton scale manufacturing without compromising purity or safety.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Chloro-6-Trifluoromethylpyridazine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthetic routes in the competitive landscape of fine chemical manufacturing. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovations like the CN102532036A trifluoromethylation process are translated into reliable industrial reality. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs that verify every batch meets the highest international standards. We understand that for R&D and procurement leaders, consistency is key, and our advanced manufacturing facilities are designed to deliver high-purity pharmaceutical intermediates with unmatched reliability and technical support.
We invite you to collaborate with us to leverage this advanced technology for your specific project needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis that demonstrates exactly how switching to this optimized route can benefit your bottom line. We encourage you to contact us today to request specific COA data and route feasibility assessments tailored to your volume requirements. By partnering with NINGBO INNO PHARMCHEM, you secure not just a supplier, but a strategic ally dedicated to enhancing your supply chain efficiency and product quality.