Advanced Chiral Synthesis of Aminoacyl-Substituted L-Phenylalanine for Commercial Scale-Up
Introduction to High-Purity Chiral Amino Acid Synthesis
The pharmaceutical industry constantly demands higher purity intermediates to ensure the safety and efficacy of final drug products, particularly in the realm of peptide therapeutics and amino acid-based medicines. Patent CN114163348A discloses a groundbreaking synthesis method for aminoacyl-substituted L-phenylalanine, a critical intermediate known for its anti-aging properties and utility in treating chronic pain and muscle repair. This molecule serves as a vital building block for complex peptide drugs and cancer therapy agents, where chiral integrity is non-negotiable. The disclosed technology represents a significant leap forward by utilizing chiral raw materials to bypass the inefficiencies of traditional racemic synthesis and resolution. By optimizing reaction parameters and employing a strategic sequence of hydroxyl activation and carbonyl insertion, the method guarantees exceptional chiral optical purity exceeding 99.9 percent. 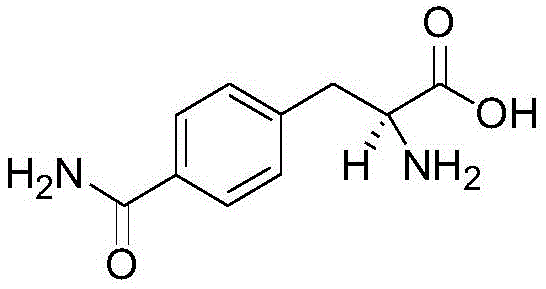 For procurement managers and R&D directors seeking a reliable pharmaceutical intermediate supplier, this patent offers a pathway to secure supply chains with superior quality metrics. The ability to produce such a complex chiral molecule without the risk of racemization addresses a major pain point in API manufacturing, ensuring that downstream products meet stringent regulatory standards for optical purity and toxicological safety.
For procurement managers and R&D directors seeking a reliable pharmaceutical intermediate supplier, this patent offers a pathway to secure supply chains with superior quality metrics. The ability to produce such a complex chiral molecule without the risk of racemization addresses a major pain point in API manufacturing, ensuring that downstream products meet stringent regulatory standards for optical purity and toxicological safety.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the preparation of aminoacyl-substituted L-phenylalanine has been plagued by significant inefficiencies inherent to achiral synthesis strategies. Conventional processes typically involve synthesizing the racemic mixture first and then attempting to separate the desired L-enantiomer from the unwanted D-enantiomer through chiral resolution. This approach suffers from a theoretical maximum yield of only 50 percent for the desired isomer, effectively wasting half of the raw materials and increasing the cost of goods sold substantially. Furthermore, the physical properties of enantiomers, such as polarity and solubility, are nearly identical, making the separation process technically challenging and often incapable of achieving the high optical purity required for medicinal use. Residual D-isomers can lead to severe toxic side effects and compromise the safety profile of the final medication. Additionally, even when starting with chiral materials, prior art methods often employed harsh reaction conditions that induced racemization during the synthesis, necessitating further purification steps that further eroded yield and increased production lead times.
The Novel Approach
In stark contrast, the novel approach detailed in the patent leverages a chirally pure starting material and maintains stereochemical integrity throughout the entire synthetic sequence. The strategy involves a sophisticated four-step cascade: hydroxyl activation, ester hydrolysis, palladium-catalyzed carbonyl insertion, and final deprotection. By carefully controlling reaction temperatures, such as cooling to -5 to 5 degrees Celsius during the activation step, the method effectively suppresses any potential racemization pathways. This allows the process to bypass the resolution step entirely, theoretically doubling the material efficiency compared to traditional racemic routes. The use of mild conditions not only preserves the chiral center but also simplifies the workup procedures, reducing the environmental footprint and operational complexity. This paradigm shift from resolution-dependent synthesis to direct chiral construction provides a robust foundation for cost reduction in API manufacturing and ensures a consistent supply of high-quality intermediates for global pharmaceutical clients.
Mechanistic Insights into Palladium-Catalyzed Carbonylation and Activation
The core of this synthetic innovation lies in the precise orchestration of organic transformations that build complexity while preserving chirality. The process initiates with the activation of the phenolic hydroxyl group on the chiral tyrosine derivative (Compound A-1) using trifluoromethanesulfonic anhydride. This conversion to a triflate (Compound A-2) creates a superb leaving group, priming the aromatic ring for subsequent nucleophilic attack or metal-catalyzed coupling. Following this, a mild alkaline hydrolysis converts the ester moiety to a carboxylic acid (Compound A-3) without disturbing the sensitive alpha-amino stereocenter. The pivotal step is the palladium-catalyzed carbonyl insertion, where carbon monoxide is inserted into the aryl-triflate bond in the presence of ammonia or an amine source. This transforms the aromatic system into the desired amide functionality found in the target glutamine-phenylalanine hybrid structure. 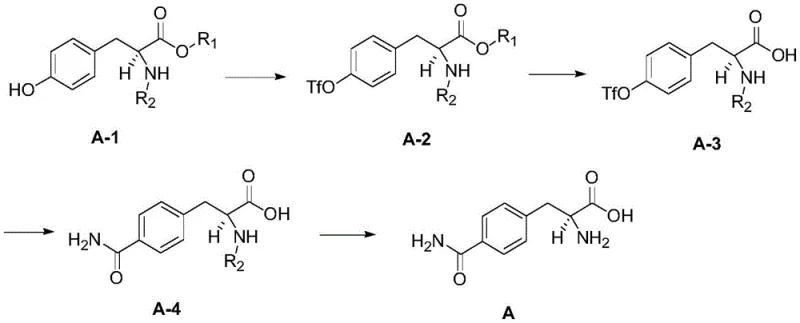 The choice of palladium catalysts, such as Pd(dppf)Cl2 or Pd(PPh3)4, along with polar aprotic solvents like DMF or DMA, is critical for facilitating this oxidative addition and migratory insertion cycle efficiently. Finally, the removal of the amino protecting group (R2), whether it be Boc, Cbz, or Fmoc, is tailored to the specific group used, ensuring the final free amine is revealed without epimerization. This mechanistic precision ensures that impurities arising from side reactions or stereochemical drift are minimized, resulting in a product profile that is exceptionally clean and suitable for direct use in sensitive peptide couplings.
The choice of palladium catalysts, such as Pd(dppf)Cl2 or Pd(PPh3)4, along with polar aprotic solvents like DMF or DMA, is critical for facilitating this oxidative addition and migratory insertion cycle efficiently. Finally, the removal of the amino protecting group (R2), whether it be Boc, Cbz, or Fmoc, is tailored to the specific group used, ensuring the final free amine is revealed without epimerization. This mechanistic precision ensures that impurities arising from side reactions or stereochemical drift are minimized, resulting in a product profile that is exceptionally clean and suitable for direct use in sensitive peptide couplings.
Impurity control is inherently built into this route through the avoidance of harsh acidic or basic conditions that typically promote racemization at the alpha-carbon. In traditional peptide synthesis, the risk of forming diketopiperazines or undergoing base-catalyzed epimerization is high. However, by performing the carbonyl insertion on a protected intermediate where the alpha-amine is shielded, the method isolates the reactive site to the aromatic ring. The hydrolysis step is conducted at room temperature with controlled pH adjustment, preventing the formation of racemates. Furthermore, the final deprotection steps are optimized for specificity; for instance, using hydrogenation for Cbz removal or mild acid for Boc removal avoids the strong bases that could scramble the chiral center. This rigorous control over the reaction environment means that the final product consistently achieves greater than 99.9 percent chiral optical purity, as verified by chiral HPLC analysis, eliminating the need for costly and yield-reducing recrystallization or chromatographic purification steps often required to meet pharmacopeial standards.
How to Synthesize Aminoacyl-Substituted L-Phenylalanine Efficiently
The synthesis protocol outlined in the patent provides a clear, scalable roadmap for producing this high-value intermediate. The procedure is designed to be operationally simple, utilizing common laboratory reagents and standard equipment, which facilitates easy technology transfer from R&D to pilot and commercial plants. The sequence begins with the activation of the phenol, followed by hydrolysis to the acid, then the key carbonylation to install the amide, and concludes with deprotection. Each step has been optimized for yield and purity, with specific molar ratios and solvent systems defined to maximize efficiency. For example, the carbonylation step utilizes a mixed solvent system of polar aprotic solvents and aqueous ammonia to ensure solubility of both the organic substrate and the gaseous reagent. This structured approach allows manufacturers to predict outcomes accurately and maintain tight quality control over the entire batch, ensuring that every kilogram produced meets the rigorous specifications required for clinical and commercial applications.
- Perform hydroxyl activation on chiral raw material A-1 using trifluoromethanesulfonic anhydride and organic base to obtain compound A-2.
- Conduct alkaline hydrolysis of compound A-2 in a water-alcohol mixed solvent to yield the carboxylic acid intermediate A-3.
- Execute palladium-catalyzed carbonyl insertion on compound A-3 using CO gas to introduce the amide functionality, forming compound A-4.
- Remove the amino protecting group from compound A-4 under specific conditions (acidic, hydrogenation, or basic) to obtain the final target product.
Commercial Advantages for Procurement and Supply Chain Teams
For supply chain leaders and procurement officers, the adoption of this synthesis method translates into tangible strategic advantages beyond mere technical superiority. The elimination of the chiral resolution step is the most significant economic driver, as it removes the inherent 50 percent yield ceiling associated with separating racemates. This fundamental shift in process chemistry leads to a drastic reduction in raw material consumption per kilogram of finished product, directly lowering the variable cost of production. Moreover, the mild reaction conditions reduce energy consumption associated with heating and cooling cycles, contributing to a more sustainable and cost-effective manufacturing profile. The use of readily available chiral starting materials ensures that supply chain bottlenecks related to exotic reagents are minimized, enhancing the reliability of supply for long-term contracts. By partnering with a supplier who utilizes this advanced methodology, pharmaceutical companies can secure a stable source of critical intermediates that are less susceptible to market volatility and raw material shortages.
- Cost Reduction in Manufacturing: The primary cost benefit arises from the atom economy and yield improvements inherent in the chiral pool approach. By avoiding the discard of the unwanted D-enantiomer, the effective yield of the process is nearly doubled compared to resolution-based methods. Additionally, the simplified purification workflow reduces the consumption of chromatography media and solvents, which are often major cost centers in fine chemical production. The avoidance of expensive transition metal scavengers, thanks to the efficient catalyst usage and workup, further drives down the cost of goods. These cumulative efficiencies allow for a more competitive pricing structure without compromising on the high purity standards demanded by regulatory bodies.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route ensures consistent batch-to-batch quality, which is crucial for maintaining uninterrupted drug production schedules. The reliance on commodity chemicals like trifluoromethanesulfonic anhydride, palladium catalysts, and common solvents means that the supply chain is not dependent on single-source specialty vendors. The scalability of the process, demonstrated by its successful execution in various solvent systems and with different protecting groups, allows manufacturers to ramp up production volume rapidly in response to market demand. This flexibility is vital for mitigating risks associated with sudden spikes in demand for oncology or pain management therapies that utilize this intermediate.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, the method offers significant advantages. The post-treatment procedures are described as green and environmentally friendly, implying reduced hazardous waste generation compared to traditional heavy metal-catalyzed or resolution-heavy processes. The ability to operate at near-ambient temperatures for several steps reduces the facility's energy load and carbon footprint. Furthermore, the high purity of the crude product minimizes the need for extensive recycling of mother liquors or complex waste treatment streams. This alignment with green chemistry principles not only simplifies regulatory compliance but also enhances the corporate sustainability profile of the manufacturing partner, a key metric for modern ESG-conscious procurement strategies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of aminoacyl-substituted L-phenylalanine. These answers are derived directly from the technical disclosures and experimental data within the patent documentation, providing clarity on the method's capabilities and limitations. Understanding these details is essential for R&D teams evaluating the feasibility of this intermediate for their specific drug candidates and for supply chain managers assessing vendor qualifications. We encourage potential partners to review these insights to fully appreciate the value proposition of this advanced synthesis technology.
Q: How does this synthesis method prevent racemization compared to traditional routes?
A: Traditional methods often start with achiral materials requiring resolution, leading to significant yield loss and potential racemization. This patented method utilizes chiral starting materials and mild reaction conditions (e.g., low temperature triflation and room temperature hydrolysis) that preserve the stereocenter, achieving over 99.9% chiral optical purity without the need for difficult chiral resolution steps.
Q: What are the key advantages for industrial scale-up of this process?
A: The process features mild operating conditions, readily available raw materials, and green post-treatment procedures. By eliminating the resolution step which typically caps yield at 50%, the overall yield is drastically improved. Furthermore, the use of standard palladium catalysis and common solvents facilitates easy adaptation to large-scale reactors.
Q: Can different amino protecting groups be used in this synthesis route?
A: Yes, the method is highly versatile regarding protecting groups. The patent explicitly demonstrates successful synthesis using Cbz, Boc, and Fmoc protecting groups (R2), with specific deprotection protocols for each (hydrogenation for Cbz, acid for Boc, base for Fmoc), allowing flexibility based on downstream peptide synthesis requirements.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Aminoacyl-Substituted L-Phenylalanine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-purity chiral intermediates play in the development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering aminoacyl-substituted L-phenylalanine with stringent purity specifications, leveraging our rigorous QC labs to verify chiral optical purity and impurity profiles for every batch. Our capability to implement the advanced synthesis methods described in patent CN114163348A positions us as a strategic partner capable of meeting the demanding requirements of global pharmaceutical innovators.
We invite you to engage with our technical procurement team to discuss how our manufacturing capabilities can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits of switching to our high-efficiency production route. We are prepared to provide specific COA data and route feasibility assessments to demonstrate our commitment to quality and reliability. Let us collaborate to accelerate your drug development timeline with a supply partner that prioritizes both scientific excellence and commercial viability.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →