Scalable Synthesis of 5-Trifluoromethyl-2'-Deoxyuridine for Global Antiviral Drug Production
The pharmaceutical industry continuously seeks robust synthetic pathways for critical antiviral agents, particularly nucleoside analogs that serve as the backbone for treating herpes simplex virus infections and other DNA viral pathogens. Patent CN100334100C discloses a novel and environmentally benign method for synthesizing 5-trifluoromethyl-2'-deoxyuridine, also known as Trifluridine, which represents a significant advancement over prior art technologies. This specific chemical entity functions as a thymidine analogue that effectively interferes with DNA synthesis, making it indispensable for clinical applications in ophthalmology and potential systemic antiviral therapies. The disclosed methodology addresses historical challenges associated with toxicity and complexity by utilizing 2'-deoxyuridine as a readily accessible starting material, thereby streamlining the entire production workflow. By shifting away from hazardous reagents traditionally employed in fluorination chemistry, this patent outlines a route that aligns perfectly with modern green chemistry principles while maintaining high structural fidelity. For global procurement teams and R&D directors, understanding the nuances of this synthesis is crucial for securing a reliable pharmaceutical intermediates supplier capable of delivering consistent quality at scale.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fluorinated nucleosides has been plagued by the reliance on aggressive and toxic reagents that pose severe safety and environmental hazards during manufacturing. Prior art methods, such as those referenced in earlier patent applications like 92102448.7, often necessitated the use of tin tetrachloride as a Lewis acid catalyst, which introduces significant complications in waste management and product purification. The presence of tin residues requires extensive and costly downstream processing to meet stringent regulatory limits for heavy metals in active pharmaceutical ingredients. Furthermore, conventional routes frequently suffered from poor regioselectivity, leading to complex impurity profiles that were difficult to separate without sacrificing overall yield. These inefficiencies not only escalated the cost reduction in antiviral agent manufacturing but also created bottlenecks in the supply chain due to the specialized handling requirements for hazardous chemicals. The environmental footprint of such processes is substantial, often generating large volumes of acidic waste streams that require neutralization and treatment before disposal, adding another layer of operational expense and compliance risk for producers.
The Novel Approach
In stark contrast, the methodology detailed in CN100334100C introduces a streamlined sequence that prioritizes safety, simplicity, and scalability without compromising chemical efficiency. The core innovation lies in the strategic application of Ceric Ammonium Nitrate (CAN) for the iodination step, which serves as a mild yet effective oxidant that avoids the generation of toxic heavy metal byproducts. This novel approach facilitates a cleaner reaction profile where the introduction of the trifluoromethyl group is achieved through a copper-mediated coupling with trifluoroiodomethane, a reagent that is easier to handle and source than many alternative fluorinating agents. The synthetic route is designed to maximize atom economy and minimize the number of isolation steps, which directly translates to enhanced supply chain reliability and reduced lead time for high-purity pharmaceutical intermediates. By employing standard protecting group strategies using acetic anhydride and trityl chloride, the process ensures that the sensitive glycosidic bond remains intact throughout the harsh conditions required for halogenation and fluorination. This robustness makes the commercial scale-up of complex nucleoside analogs far more feasible for contract development and manufacturing organizations aiming to expand their portfolio of antiviral solutions.
Mechanistic Insights into CAN-Mediated Iodination and Trifluoromethylation
The chemical elegance of this synthesis is rooted in the precise control of reactivity at the C-5 position of the uracil ring, which is achieved through a radical-mediated iodination mechanism driven by the CAN reagent system. In this critical transformation, the cerium(IV) species acts as a single-electron oxidant that generates iodine radicals from molecular iodine, which then selectively attack the electron-rich C-5 position of the protected pyrimidine base. This regioselective functionalization is paramount because it sets the stage for the subsequent cross-coupling reaction, ensuring that the trifluoromethyl group is installed exactly where it is needed to confer biological activity. The use of acetonitrile as a solvent further stabilizes the radical intermediates, preventing unwanted side reactions at the sugar moiety or the nitrogen atoms of the base. Understanding this mechanistic pathway allows process chemists to fine-tune reaction parameters such as temperature and stoichiometry to optimize the conversion rate, which is reported to be highly efficient in the provided embodiments. The ability to control the oxidation state of the reaction mixture prevents over-oxidation of the sugar hydroxyls, preserving the integrity of the nucleoside scaffold which is essential for the final drug's efficacy.
Following the iodination, the introduction of the trifluoromethyl group proceeds via a copper-catalyzed substitution that replaces the iodine atom with the CF3 moiety, a transformation that is thermodynamically favorable yet kinetically challenging without the correct catalytic system. The patent specifies the use of copper powder in HMPA at elevated temperatures, which facilitates the formation of a trifluoromethyl-copper species that undergoes transmetallation with the aryl-iodide intermediate. This step is critical for establishing the metabolic stability of the final drug candidate, as the carbon-fluorine bond is one of the strongest in organic chemistry and significantly alters the pharmacokinetic properties of the molecule. Subsequent deprotection using mild acid hydrolysis removes the acetyl and trityl groups without cleaving the glycosidic bond, a common pitfall in nucleoside chemistry that this route successfully avoids. 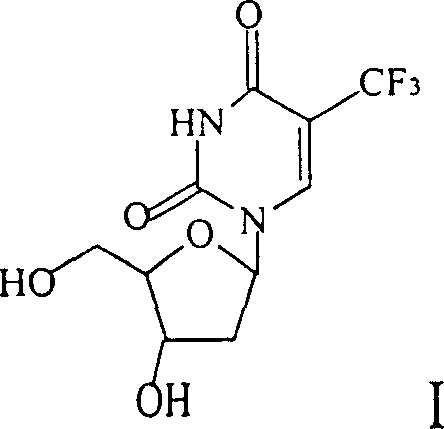 The final structure, as depicted in the chemical diagram, confirms the successful installation of the trifluoromethyl group while maintaining the stereochemistry of the deoxyribose sugar, which is vital for recognition by viral polymerases. This level of structural precision ensures that the resulting intermediate meets the rigorous purity specifications required for downstream formulation into antiviral medications.
The final structure, as depicted in the chemical diagram, confirms the successful installation of the trifluoromethyl group while maintaining the stereochemistry of the deoxyribose sugar, which is vital for recognition by viral polymerases. This level of structural precision ensures that the resulting intermediate meets the rigorous purity specifications required for downstream formulation into antiviral medications.
How to Synthesize 5-Trifluoromethyl-2'-Deoxyuridine Efficiently
Executing this synthesis requires careful attention to the sequence of protection and deprotection steps to ensure maximum yield and minimal impurity formation throughout the process. The protocol begins with the acetylation of 2'-deoxyuridine to protect the primary and secondary hydroxyl groups, followed by the critical iodination step which must be monitored closely to prevent di-iodination or degradation. Detailed standardized synthetic steps see the guide below which outlines the specific molar ratios, solvent choices, and workup procedures necessary to replicate the high yields reported in the patent literature. Process engineers should note that the removal of copper residues after the trifluoromethylation step may require specific chelating agents or filtration techniques to ensure the final product is free from metal contaminants. Adhering to these operational guidelines is essential for achieving the commercial scale-up of complex nucleoside analogs while maintaining compliance with Good Manufacturing Practices (GMP).
- Protect the 3' and 5' hydroxyl groups of 2'-deoxyuridine using acetic anhydride to form 3',5'-di-acetyl-2'-deoxyuridine.
- Perform iodination at the 5-position using Iodine and Ceric Ammonium Nitrate (CAN) in acetonitrile to yield 5-iodo-3',5'-di-acetyl-2'-deoxyuridine.
- Execute trifluoromethylation using CF3I and copper powder, followed by acid hydrolysis to remove protecting groups and obtain the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of this synthetic route offers profound benefits that extend beyond mere chemical feasibility, directly impacting the bottom line and operational resilience of pharmaceutical manufacturers. The elimination of tin-based reagents removes a major regulatory hurdle, as the testing and validation for heavy metal clearance are resource-intensive processes that can delay product launch timelines. By utilizing a pathway that relies on more benign oxidants and catalysts, companies can significantly reduce the environmental compliance burden and lower the costs associated with hazardous waste disposal and treatment facilities. This shift towards greener chemistry not only aligns with corporate sustainability goals but also mitigates the risk of supply chain disruptions caused by tightening environmental regulations on chemical manufacturing. Furthermore, the use of 2'-deoxyuridine as a starting material leverages an existing, mature supply chain for nucleosides, ensuring that raw material availability remains stable even during periods of market volatility. These factors combined create a compelling value proposition for procurement managers seeking to optimize their vendor portfolio with partners who prioritize both efficiency and responsibility.
- Cost Reduction in Manufacturing: The streamlined nature of this synthesis eliminates multiple purification stages that are typically required to remove toxic metal residues, leading to substantial cost savings in both materials and labor. By avoiding the use of expensive and hazardous Lewis acids like tin tetrachloride, the process reduces the need for specialized corrosion-resistant equipment and complex quenching protocols. The high yields observed in the intermediate steps, such as the 89% yield in the trifluoromethylation reaction, contribute to a more efficient use of raw materials, minimizing waste and maximizing output per batch. Additionally, the simplified workup procedures reduce the consumption of solvents and energy, further driving down the overall cost of goods sold for this critical antiviral intermediate. These efficiencies allow for a more competitive pricing structure without compromising on the quality or purity of the final product.
- Enhanced Supply Chain Reliability: The reliance on commercially available reagents such as iodine, acetic anhydride, and copper powder ensures that the production of this intermediate is not bottlenecked by the scarcity of exotic chemicals. This accessibility translates to reduced lead time for high-purity pharmaceutical intermediates, allowing manufacturers to respond more quickly to fluctuations in demand for antiviral medications. The robustness of the reaction conditions means that the process is less susceptible to minor variations in raw material quality, providing a consistent and reliable output that is crucial for maintaining continuous supply to downstream formulators. Moreover, the reduced environmental impact of the process minimizes the risk of regulatory shutdowns or fines, ensuring long-term continuity of supply for global healthcare markets. This stability is a key differentiator for supply chain heads looking to de-risk their procurement strategies for essential medicines.
- Scalability and Environmental Compliance: The synthetic route is inherently designed for scalability, with reaction conditions that can be safely translated from laboratory glassware to large-scale industrial reactors without significant re-engineering. The absence of highly exothermic or dangerous steps reduces the safety risks associated with large-batch production, facilitating smoother technology transfer and faster ramp-up times. From an environmental standpoint, the generation of less hazardous waste simplifies the permitting process for new manufacturing sites and supports the company's commitment to sustainable operations. The ability to produce this compound with a lower ecological footprint enhances the brand reputation of the manufacturer and aligns with the increasing demand for green pharmaceuticals from healthcare providers and patients alike. This combination of scalability and compliance makes the technology a future-proof asset for any organization invested in the antiviral sector.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of 5-trifluoromethyl-2'-deoxyuridine, based on the detailed specifications found in the underlying patent documentation. These insights are intended to clarify the operational advantages and chemical characteristics of this specific synthetic route for stakeholders evaluating its potential for integration into their supply chains. Understanding these details is essential for making informed decisions about sourcing strategies and process development investments in the competitive landscape of antiviral drug manufacturing.
Q: What are the advantages of the CAN-mediated iodination method over traditional tin-based methods?
A: The use of Ceric Ammonium Nitrate (CAN) eliminates the need for toxic tin tetrachloride, significantly reducing environmental pollution and simplifying the downstream purification process by avoiding heavy metal residue removal.
Q: Is the starting material 2'-deoxyuridine readily available for commercial scale-up?
A: Yes, 2'-deoxyuridine is a commercially accessible nucleoside base, which ensures a stable supply chain and reduces raw material procurement risks compared to exotic starting materials.
Q: How does this synthesis route impact the purity profile of the final antiviral intermediate?
A: The stepwise protection and deprotection strategy allows for precise control over regioselectivity, minimizing side reactions and ensuring a high-purity impurity profile suitable for strict pharmaceutical standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-2'-Deoxyuridine Supplier
At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure required to translate complex laboratory patents into robust, commercial-scale manufacturing processes that meet the highest global standards. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can support your volume requirements whether you are in the clinical trial phase or full-scale market launch. We understand that the purity of nucleoside analogs is critical for patient safety, which is why our facilities are equipped with rigorous QC labs and advanced analytical instrumentation to verify stringent purity specifications for every batch we release. Our commitment to quality assurance extends beyond simple compliance; we actively engage in process optimization to enhance yield and reduce impurities, delivering a superior product that accelerates your drug development timeline.
We invite you to collaborate with us to leverage this advanced synthetic technology for your antiviral pipeline, benefiting from our deep understanding of fluorinated nucleoside chemistry and supply chain dynamics. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume needs and project timelines. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can add tangible value to your organization. Partnering with us means securing a dependable source of high-quality intermediates that will empower your research and commercial success in the therapeutic market.