Advanced Synthesis of 5-Formylpyrimidine Carbocyclic Nucleosides for Antiviral Drug Development
Advanced Synthesis of 5-Formylpyrimidine Carbocyclic Nucleosides for Antiviral Drug Development
The pharmaceutical landscape for antiviral therapeutics has been profoundly shaped by the evolution of nucleoside analogs, particularly those featuring carbocyclic structures that offer superior metabolic stability. Patent CN102603652A introduces a pivotal advancement in this domain by disclosing a robust preparation method for 5-formylpyrimidine carbocyclic nucleosides. These compounds are not merely end-products but serve as critical, high-value intermediates for synthesizing more complex 5-substituted pyrimidine carbocyclic nucleoside drugs. The strategic introduction of a formyl group at the 5-position of the pyrimidine base unlocks a myriad of synthetic possibilities, allowing medicinal chemists to construct diverse libraries of potential antiviral and antitumor agents. This patent represents a significant leap forward for any organization seeking a reliable pharmaceutical intermediate supplier capable of delivering complex nucleoside scaffolds with precision and efficiency.
The significance of this technology is underscored by the clinical success of existing carbocyclic nucleosides such as Carbovir, Abacavir, and Entecavir, which have set the benchmark for anti-HIV and anti-HBV therapies. 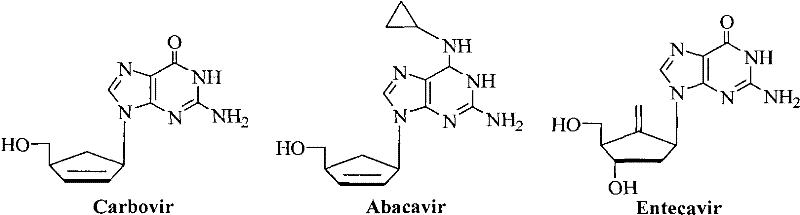 As illustrated by these market-leading drugs, the replacement of the natural sugar moiety with a carbocyclic ring confers resistance to enzymatic degradation, thereby prolonging the drug's half-life and efficacy. The methodology described in CN102603652A builds upon this foundation by providing a streamlined pathway to functionalized precursors. By establishing a reliable source for these 5-formyl derivatives, manufacturers can accelerate the discovery pipeline for next-generation antivirals, addressing the urgent global need for treatments with improved resistance profiles and reduced toxicity.
As illustrated by these market-leading drugs, the replacement of the natural sugar moiety with a carbocyclic ring confers resistance to enzymatic degradation, thereby prolonging the drug's half-life and efficacy. The methodology described in CN102603652A builds upon this foundation by providing a streamlined pathway to functionalized precursors. By establishing a reliable source for these 5-formyl derivatives, manufacturers can accelerate the discovery pipeline for next-generation antivirals, addressing the urgent global need for treatments with improved resistance profiles and reduced toxicity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the functionalization of the pyrimidine base in nucleoside analogs has been fraught with challenges, particularly when attempting to introduce reactive groups like aldehydes without compromising the integrity of the sensitive sugar or carbocyclic ring. Conventional oxidation methods often rely on harsh reagents or extreme conditions that can lead to over-oxidation, producing carboxylic acids instead of the desired aldehyde, or causing degradation of the nucleobase. Furthermore, traditional routes may require multiple protection and deprotection steps that are labor-intensive and result in poor overall yields. The instability of the glycosidic bond in natural nucleosides also poses a significant risk during aggressive chemical transformations, leading to the formation of free base impurities that are difficult to remove. These limitations have historically created bottlenecks in the cost reduction in API manufacturing, as low yields and complex purification processes drive up production costs and extend lead times.
The Novel Approach
The methodology outlined in patent CN102603652A offers a transformative solution by employing a mild, three-step sequence that prioritizes selectivity and operational simplicity. The core innovation lies in the specific oxidation system utilized to convert the 5-methyl group directly to a 5-formyl group under controlled alkaline conditions. 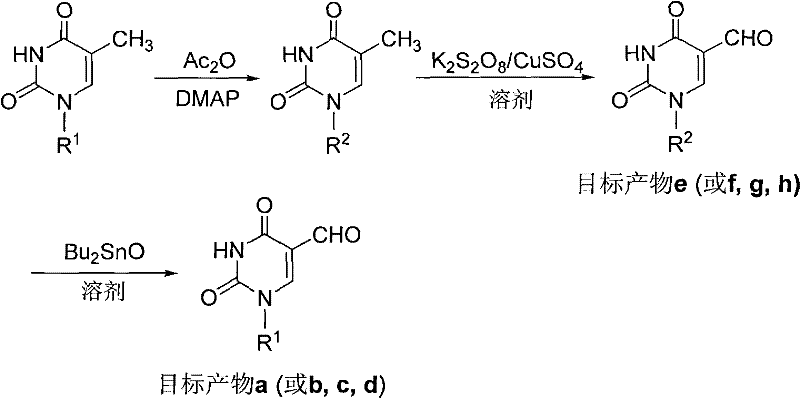 This approach avoids the pitfalls of over-oxidation and preserves the stereochemical integrity of the carbocyclic ring. By starting with readily available 5-methylpyrimidine carbocyclic nucleosides, the process eliminates the need for constructing the base-ring connection from scratch, thereby shortening the synthetic timeline. The use of acetic anhydride for protection and dibutyltin oxide for deprotection ensures that the reactive aldehyde functionality is only exposed in the final step, minimizing side reactions. This novel approach effectively resolves the scalability issues associated with previous methods, making it an ideal candidate for the commercial scale-up of complex pharmaceutical intermediates.
This approach avoids the pitfalls of over-oxidation and preserves the stereochemical integrity of the carbocyclic ring. By starting with readily available 5-methylpyrimidine carbocyclic nucleosides, the process eliminates the need for constructing the base-ring connection from scratch, thereby shortening the synthetic timeline. The use of acetic anhydride for protection and dibutyltin oxide for deprotection ensures that the reactive aldehyde functionality is only exposed in the final step, minimizing side reactions. This novel approach effectively resolves the scalability issues associated with previous methods, making it an ideal candidate for the commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Potassium Persulfate-Copper Sulfate Oxidation
The heart of this synthetic strategy is the second step, where the methyl group at the 5-position of the uracil ring is oxidized to an aldehyde. This transformation is achieved using a radical oxidation system comprising potassium persulfate (K2S2O8) and copper sulfate (CuSO4) in a mixed solvent of acetonitrile and water. The mechanism likely involves the generation of sulfate radical anions from the persulfate, which are activated by the copper catalyst to abstract hydrogen atoms from the benzylic-like methyl position. The presence of a base, such as 2,6-lutidine or pyridine, is crucial for neutralizing the acidic byproducts and facilitating the formation of the carbonyl group. This specific catalytic system is highly advantageous because it operates at a moderate temperature range of 60-70°C, which is sufficiently energetic to drive the reaction to completion without inducing thermal decomposition of the nucleoside scaffold. The selectivity of this oxidation is paramount, as it must stop at the aldehyde stage rather than proceeding to the carboxylic acid, a feat accomplished by the precise control of reaction time and stoichiometry described in the patent.
Equally important is the role of the protecting groups in managing the reactivity of the molecule throughout the synthesis. The initial acetylation of the hydroxyl groups on the carbocyclic ring serves a dual purpose: it increases the lipophilicity of the intermediate, improving its solubility in the organic oxidation medium, and it prevents the oxidation of the alcohol functionalities themselves. The subsequent deprotection using dibutyltin oxide (Bu2SnO) is a sophisticated choice for a transesterification reaction. Unlike harsh acidic or basic hydrolysis which might attack the newly formed aldehyde or the glycosidic bond, the tin-mediated deprotection proceeds under neutral to mildly basic reflux conditions. This gentle removal of the acetyl groups ensures that the final high-purity carbocyclic nucleoside is obtained with minimal formation of degradation impurities. The ability to control the impurity profile through such mechanistic finesse is a key consideration for R&D directors focused on regulatory compliance and product quality.
How to Synthesize 5-Formyl-2'-Deoxyuridine Carbocyclic Nucleoside Efficiently
The practical execution of this synthesis is designed to be accessible for industrial application, utilizing common reagents and standard equipment. The process begins with the protection of the starting material, followed by the critical oxidation step, and concludes with the removal of the protecting groups to reveal the active pharmacophore. Each step is monitored via TLC to ensure optimal conversion before proceeding, which is a best practice for maintaining batch consistency. The patent provides specific examples, such as the synthesis of 5-formyl-2'-deoxyuridine carbocyclic nucleoside (Target Product a), demonstrating the viability of the route with concrete experimental data. For teams looking to implement this chemistry, understanding the nuances of workup and purification is essential to achieving the reported yields. The detailed standardized synthesis steps are provided in the guide below to facilitate immediate technology transfer and process validation.
- Hydroxyl Protection: React 5-methyl-2'-deoxyuridine carbocyclic nucleoside with acetic anhydride and DMAP catalyst at room temperature to obtain the acetyl-protected intermediate.
- Methyl Oxidation: Oxidize the protected intermediate using a potassium persulfate and copper sulfate system in acetonitrile/water at 60-70°C to introduce the formyl group.
- Deprotection: Remove the acetyl protecting groups by refluxing the oxidized product with dibutyltin oxide in a suitable solvent like methanol to yield the final target compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of the synthesis route described in CN102603652A presents compelling economic and logistical benefits. The primary advantage stems from the simplification of the process flow; by reducing the number of unit operations and avoiding exotic reagents, the overall manufacturing footprint is significantly reduced. The use of commodity chemicals like acetic anhydride, potassium persulfate, and copper sulfate ensures that raw material sourcing is stable and cost-effective, mitigating the risks associated with supply chain disruptions for specialized reagents. Furthermore, the mild reaction conditions translate to lower energy consumption and reduced safety hazards, which directly correlates to lower operational expenditures. This process optimization allows for a substantial cost savings in the production of these high-value intermediates, making the final antiviral APIs more economically viable to produce at scale.
- Cost Reduction in Manufacturing: The elimination of harsh reaction conditions and the use of catalytic amounts of copper sulfate significantly lower the cost of goods sold. By avoiding the need for cryogenic temperatures or high-pressure reactors, the capital expenditure required for manufacturing equipment is drastically simplified. Additionally, the high selectivity of the oxidation step reduces the burden on downstream purification processes, such as chromatography, which are often the most expensive part of nucleoside manufacturing. This efficiency gain means that resources can be allocated more effectively, driving down the unit cost of the intermediate without compromising quality.
- Enhanced Supply Chain Reliability: The reliance on widely available inorganic oxidants and common organic solvents like acetonitrile and methanol ensures a robust supply chain. Unlike processes that depend on single-source proprietary catalysts, this method allows for multi-vendor sourcing of key inputs, reducing the risk of production stoppages due to material shortages. The operational simplicity also means that the process can be easily transferred between different manufacturing sites if necessary, providing flexibility in capacity planning. This reliability is crucial for maintaining continuous supply to downstream API manufacturers who depend on just-in-time delivery models.
- Scalability and Environmental Compliance: The three-step sequence is inherently scalable, moving seamlessly from gram-scale laboratory synthesis to multi-kilogram pilot production. The waste stream generated is relatively manageable, consisting primarily of aqueous salts and organic solvents that can be treated using standard effluent protocols. The avoidance of heavy metal contaminants in the final product, thanks to the efficient deprotection and purification steps, simplifies the regulatory filing process. This environmental compatibility aligns with modern green chemistry principles, reducing the ecological footprint of pharmaceutical manufacturing and ensuring long-term sustainability.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of 5-formylpyrimidine carbocyclic nucleosides. These answers are derived directly from the technical specifications and beneficial effects detailed in the patent literature. Understanding these aspects is vital for stakeholders evaluating the feasibility of integrating this intermediate into their drug development pipelines. The clarity provided here aims to reduce lead time for high-purity pharmaceutical intermediates by pre-emptively resolving technical uncertainties.
Q: What is the primary advantage of the 5-formyl group in carbocyclic nucleosides?
A: The aldehyde carbonyl group at the 5-position possesses rich reactivity, serving as a versatile handle for further structural modifications to create novel 5-substituted pyrimidine carbocyclic nucleosides with potential antiviral and antitumor activities.
Q: Why is the carbocyclic structure preferred over natural ribose in antiviral drugs?
A: Replacing the D-ribose with a five-membered carbon ring transforms the unstable hemiaminal glycosidic linkage into a stable tertiary amine bond, significantly enhancing metabolic stability against enzymatic cleavage while maintaining biological activity.
Q: What are the key reaction conditions for the oxidation step in this patent?
A: The oxidation step utilizes a mixture of potassium persulfate and copper sulfate under alkaline conditions (using bases like 2,6-lutidine) at a moderate temperature range of 60-70°C, ensuring mild and controlled conversion of the methyl group to an aldehyde.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Formylpyrimidine Carbocyclic Nucleoside Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of antiviral therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and timeliness. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our expertise in nucleoside chemistry allows us to navigate the complexities of this synthesis route, guaranteeing a consistent supply of 5-formylpyrimidine carbocyclic nucleosides that adhere to the highest industry standards.
We invite you to collaborate with us to leverage this advanced synthesis technology for your next-generation drug candidates. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our manufacturing capabilities can accelerate your path to market while optimizing your overall production costs.