Scalable Synthesis of Scleropentaside A via Novel Pd-Cu Catalyzed Desulfurization
The pharmaceutical industry continuously seeks robust synthetic pathways for bioactive natural products, particularly those with complex stereochemistry like Scleropentaside A. This compound, isolated from plants such as Scleropyrum pentandrum, exhibits significant potential in traditional medicine for treating rheumatic pain and skin conditions, yet its chemical synthesis has historically been plagued by low efficiency and hazardous reagents. A breakthrough detailed in Chinese patent CN112409344B introduces a streamlined four-step methodology that drastically enhances overall throughput while prioritizing operator safety and environmental compliance. By shifting from traditional benzyl protection strategies to a novel thioester-mediated cross-coupling approach, this technology addresses the critical bottlenecks of yield and scalability that have long hindered the commercial availability of high-purity Scleropentaside A. For R&D directors and procurement specialists, understanding this shift is vital for securing a reliable supply chain for this valuable pharmaceutical intermediate.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the total synthesis of Scleropentaside A has been characterized by inefficient multi-step sequences that suffer from substantial material loss at every stage. Early attempts, such as the perbenzyl-protected route reported by Walczak et al., relied on harsh conditions that resulted in a meager overall yield of merely 40%, rendering the process economically unviable for industrial application. Subsequent improvements, including the beta-selective C-glycosylation strategy published by Boehlich and Schützenmeister, managed to slightly increase efficiency but still capped the total yield at approximately 47% over four to five steps. These conventional pathways often necessitate the use of toxic oxidants like iodine and hydrogen peroxide for deprotection, introducing severe safety hazards and complicating waste management protocols. Furthermore, the instability of intermediates under standard basic deprotection conditions frequently leads to the formation of elimination by-products, necessitating costly and time-consuming purification efforts that erode profit margins.
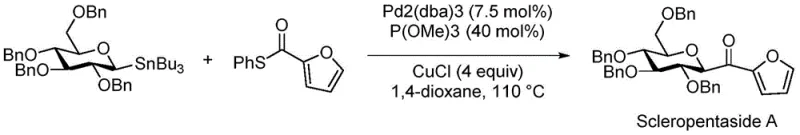
The Novel Approach
In stark contrast to these legacy methods, the innovative process outlined in patent CN112409344B leverages a highly efficient thioester coupling strategy to achieve a total yield exceeding 69%, representing a paradigm shift in synthetic efficiency. The core of this advancement lies in the conversion of a glycosyl acid into a thioester intermediate, which then undergoes a palladium-copper catalyzed coupling with 2-furanboronic acid to install the furan moiety with exceptional fidelity. This route not only simplifies the step count but also operates under milder conditions that preserve the delicate stereochemical integrity of the sugar backbone. By utilizing benign solvents like dichloromethane and tetrahydrofuran, and avoiding hazardous halogenated oxidants, the new method aligns perfectly with modern green chemistry principles. This technological leap ensures that manufacturers can produce gram-level quantities with consistent quality, directly addressing the supply continuity concerns of global procurement teams.
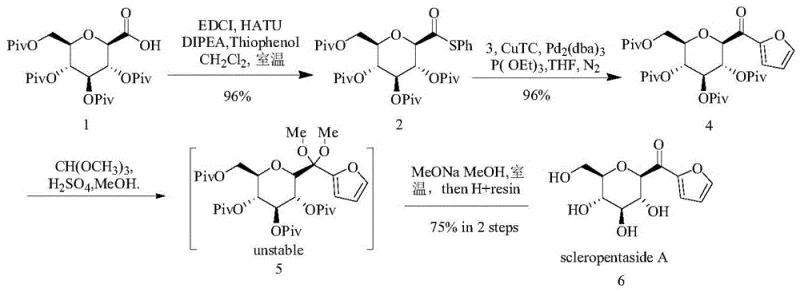
Mechanistic Insights into Pd-Cu Catalyzed Desulfurization and Impurity Control
The mechanistic elegance of this synthesis is anchored in the dual-catalyst system employing Pd2(dba)3 and CuTC, which facilitates a seamless coupling-desulfurization reaction. In this transformation, the thioester acts as a superior electrophile compared to traditional halides, allowing for the rapid formation of the C-C bond at the anomeric position without epimerization. The copper co-catalyst plays a pivotal role in activating the boronic acid and facilitating the transmetallation step, ensuring that the reaction proceeds to completion with yields consistently above 96% for this specific transformation. This high level of conversion minimizes the burden on downstream purification, as the crude reaction mixture contains significantly fewer side products. For process chemists, this means a more predictable impurity profile and a reduced risk of carryover contaminants that could compromise the final API quality.
Perhaps the most critical innovation in this patent is the strategic handling of the deprotection phase to prevent the formation of elimination by-product 7. Conventional basic hydrolysis of the pivaloyl groups on the furanone intermediate typically triggers an unwanted elimination reaction due to the acidity of the alpha-proton adjacent to the carbonyl. To circumvent this, the inventors introduced a transient carbonyl protection step, converting the ketone into a dimethyl ketal using trimethyl orthoformate and methanol. This temporary masking reduces the electron-withdrawing nature of the carbonyl, thereby suppressing the elimination pathway during the subsequent saponification with sodium methoxide. As illustrated in the comparative reaction scheme, this subtle modification shifts the product distribution almost entirely towards the desired Scleropentaside A, effectively eliminating the major impurity that plagued previous attempts.
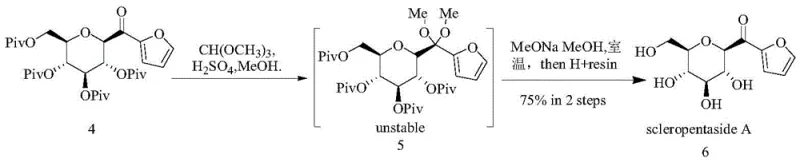
How to Synthesize Scleropentaside A Efficiently
Implementing this synthesis requires precise control over reaction parameters, particularly during the esterification and coupling stages where moisture sensitivity can impact catalyst performance. The process begins with the activation of the glycosyl acid using HATU and EDCI in the presence of DIPEA, followed by the addition of thiophenol to generate the thioester precursor in near-quantitative yield. The subsequent coupling step demands an inert nitrogen atmosphere to protect the palladium catalyst from oxidation, with reaction times optimized between 6 to 16 hours to ensure full consumption of the starting material. While the detailed stoichiometry and workup procedures are critical for reproducibility, the overarching workflow is designed to be robust enough for transfer from laboratory bench to pilot plant reactors.
- Perform esterification of glycosyl acid with thiophenol using HATU and DIPEA in dichloromethane to form the thioester intermediate.
- Execute a coupling-desulfurization reaction between the thioester and 2-furanboronic acid using Pd2(dba)3 and CuTC catalysts.
- Protect the carbonyl group as a ketal using methanol and trimethyl orthoformate, followed by deprotection with sodium methoxide and acidic resin.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers tangible benefits that extend far beyond simple yield metrics. The primary advantage lies in the drastic simplification of the raw material portfolio; by replacing scarce or hazardous reagents with commodity chemicals like thiophenol and 2-furanboronic acid, the supply chain becomes significantly more resilient to market fluctuations. The elimination of toxic iodine and peroxide reagents not only reduces the cost of hazardous waste disposal but also lowers the regulatory burden associated with handling dangerous goods, facilitating smoother logistics and storage. Furthermore, the high selectivity of the Pd-Cu catalytic system means that less solvent and stationary phase are required for purification, directly translating to lower operational expenditures per kilogram of finished product.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the exceptional step-wise yields, which exceed 96% for the key coupling and esterification transformations. By minimizing material loss at each stage, the overall consumption of expensive chiral starting materials is significantly reduced, leading to a lower cost of goods sold. Additionally, the avoidance of complex chromatographic separations required to remove elimination by-products streamlines the manufacturing timeline, allowing for higher throughput in existing facilities without the need for capital-intensive equipment upgrades.
- Enhanced Supply Chain Reliability: The reliance on stable, commercially available reagents ensures that production schedules are not disrupted by the lead times associated with custom-synthesized intermediates. The robustness of the thioester intermediate allows for potential isolation and storage, providing a buffer against upstream supply variability. This stability is crucial for maintaining continuous production runs, ensuring that downstream customers receive their orders of high-purity pharmaceutical intermediates without delay, thereby strengthening long-term supplier relationships.
- Scalability and Environmental Compliance: From an environmental perspective, the substitution of halogenated oxidants with methanol-based deprotection aligns with increasingly stringent global emissions standards. The process generates less hazardous waste, simplifying the permitting process for scale-up to multi-kilogram or tonne-level production. The use of common solvents like dichloromethane and tetrahydrofuran, which are easily recovered and recycled in modern plants, further enhances the sustainability profile of the manufacturing operation, making it an attractive option for companies committed to green chemistry initiatives.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on yield expectations, impurity profiles, and safety considerations. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: What is the total yield improvement of this new synthesis method?
A: The new method described in patent CN112409344B achieves a total yield of over 69%, which is a significant improvement compared to previous methods that yielded only 40% to 47%.
Q: How does this process avoid the formation of elimination by-products?
A: The process utilizes a unique carbonyl protection strategy where the ketone is converted to a ketal intermediate before deprotection. This reduces the elimination activity of the vicinal hydroxyl group, preventing the formation of the unwanted alkene by-product.
Q: Are the reagents used in this synthesis safe for large-scale production?
A: Yes, the method replaces toxic iodine and hydrogen peroxide reagents commonly used in prior art with safer, non-toxic alternatives like methanol and acidic resins, making it more suitable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Scleropentaside A Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of accessing high-quality natural product derivatives for drug discovery and development. Our team of expert process chemists has thoroughly analyzed the methodology described in CN112409344B and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We are equipped with rigorous QC labs and state-of-the-art analytical instrumentation to ensure that every batch of Scleropentaside A meets stringent purity specifications, free from the elimination by-products that characterize inferior synthesis routes. Our commitment to technical excellence ensures that your R&D projects proceed without the interruption of supply shortages or quality deviations.
We invite you to collaborate with us to leverage this advanced synthetic technology for your specific applications. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating exactly how this high-yield route can optimize your budget. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments, ensuring that your transition to this superior manufacturing process is seamless, efficient, and commercially advantageous.