Advanced Configuration Conversion Technology for High-Purity Toremifene Manufacturing
The pharmaceutical industry constantly seeks robust synthetic pathways that maximize yield while minimizing environmental impact, particularly for high-value antitumor agents like Toremifene. A pivotal advancement in this domain is documented in patent CN102126969A, which discloses a method for realizing high-stereoselectivity synthesis of Toremifene through a novel configuration conversion strategy. This technology addresses the longstanding challenge of isolating the biologically active Z-isomer from the inactive E-isomer during the synthesis of triaryl butenol intermediates. By leveraging the differential solubility of isomeric hydrochloride salts, the process drives the chemical equilibrium towards the desired product, effectively overcoming the thermodynamic limitations of conventional acid-catalyzed elimination reactions. For global procurement teams and R&D directors, understanding this mechanism is crucial for securing a reliable pharmaceutical intermediates supplier capable of delivering consistent quality. The implications of this technology extend beyond mere academic interest, offering tangible benefits in cost reduction in pharmaceutical intermediates manufacturing and enhancing the overall stability of the supply chain for critical oncology medications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical synthetic routes for Toremifene precursors have been fraught with inefficiencies that significantly hamper commercial viability and increase production costs. Early methodologies, such as those reported by R.J. Toivola, relied on acidic alcohol elimination which typically resulted in a Z/E isomer ratio of approximately 2:1, necessitating laborious stepwise crystallization to isolate the pure Z-configuration. This approach not only capped the theoretical yield at a mere 41% but also generated substantial chemical waste due to the discard of the E-isomer rich mother liquors. Furthermore, alternative attempts using concentrated hydrochloric acid at elevated temperatures often favored the formation of the E-isomer or resulted in complex mixtures requiring extensive purification. The reliance on repeated recrystallization steps not only extends the production lead time but also introduces risks of product loss and variability in purity profiles. For a procurement manager, these inefficiencies translate directly into higher raw material consumption and unpredictable delivery schedules, making the search for high-purity pharmaceutical intermediates increasingly challenging in a competitive market.
The Novel Approach
In stark contrast to these legacy methods, the innovative process described in the patent utilizes a dynamic equilibrium shift driven by selective crystallization to achieve superior stereoselectivity. Instead of accepting the thermodynamic equilibrium of the solution, this method actively removes the Z-type triaryl butenol hydrochloride from the reaction mixture as it forms. By maintaining the reaction in a heterogeneous system using concentrated hydrochloric acid, the Z-isomer precipitates as a solid while the E-isomer remains dissolved in the mother liquor. This continuous removal disrupts the equilibrium, forcing the remaining E-isomer to convert into the Z-form to restore balance, thereby driving the reaction towards near-completion. The result is a dramatic improvement in total recovery, with yields reaching upwards of 85% to 96% from the triaryl butyleneglycol starting material. This streamlined approach eliminates the need for multiple recrystallization cycles, simplifying the workflow and ensuring a more consistent supply of commercial scale-up of complex pharmaceutical intermediates.
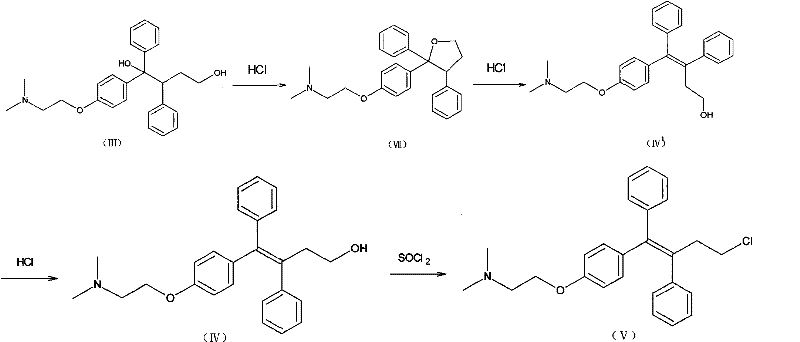
Mechanistic Insights into Acid-Catalyzed Configuration Conversion
The core of this technological breakthrough lies in the intricate interplay between acid catalysis and solubility dynamics within the reaction medium. When triaryl butyleneglycol is suspended in concentrated hydrochloric acid, it undergoes an elimination reaction to form triaryl butenol, which exists in both Z and E configurations. Under standard homogeneous conditions, the equilibrium heavily favors the E-isomer, often resulting in ratios as skewed as 9:1 against the desired product. However, the patent reveals that the hydrochloride salt of the Z-isomer possesses significantly lower solubility in the concentrated acid medium compared to its E-counterpart. By carefully controlling the temperature, typically cooling the mixture to between 10°C and 45°C after an initial heating phase, the Z-isomer hydrochloride reaches its saturation point and crystallizes out of the solution. This physical separation is the key driver that allows the chemical conversion to proceed far beyond normal equilibrium limits, effectively acting as a continuous purification and synthesis loop.
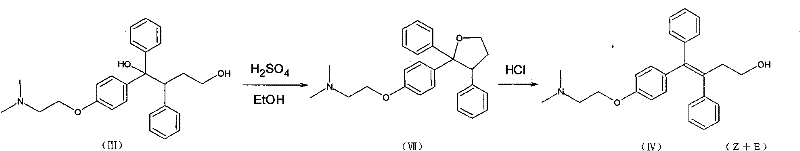
Furthermore, the mechanism includes a robust impurity control strategy that ensures the final product meets stringent medicinal standards without excessive downstream processing. The process inherently minimizes the formation of cyclization by-products, such as the tetrahydrofuran derivative, by optimizing the acid concentration and reaction time. Monitoring via TLC indicates that while cyclization intermediates may form transiently, the conditions favor their conversion back to the butenol species or their exclusion from the crystallizing Z-isomer lattice. The subsequent chlorination step using thionyl chloride is performed on the isolated, high-purity Z-isomer hydrochloride, which prevents the propagation of stereochemical impurities into the final Toremifene molecule. This level of control over the impurity profile is critical for R&D directors who must ensure that the API intermediate does not introduce toxicological risks or complicate the regulatory filing process for the final drug product.
How to Synthesize Toremifene Efficiently
The practical implementation of this synthesis route requires precise adherence to the specified reaction parameters to maximize the benefits of the configuration conversion mechanism. Operators must begin by suspending the triaryl butyleneglycol precursor in a sufficient volume of concentrated hydrochloric acid, ensuring the acid-to-substrate ratio is high enough to facilitate the elimination reaction without diluting the system excessively. The mixture is then subjected to a controlled thermal cycle, initially heating to dissolve the reactants and initiate the conversion, followed by a gradual cooling phase to induce the critical crystallization of the Z-isomer hydrochloride. Detailed standardized synthesis steps see the guide below.
- Suspend triaryl butyleneglycol in concentrated hydrochloric acid and heat to initiate elimination and cyclization reactions.
- Cool the reaction mixture to induce crystallization of the Z-type triaryl butenol hydrochloride, separating it from the E-type isomer in the mother liquor.
- Alkalize the isolated solid and perform chlorination with thionyl chloride to obtain the final Toremifene product with high purity.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this stereoselective synthesis method offers profound advantages that resonate deeply with the strategic goals of procurement and supply chain leadership. The primary benefit stems from the substantial increase in reaction yield, which directly correlates to a reduction in the cost of goods sold by maximizing the output from every kilogram of raw material purchased. Unlike traditional methods that discard significant portions of the reaction mass as waste or mother liquor, this process recovers the majority of the substrate as valuable product, thereby optimizing resource utilization. Additionally, the simplification of the purification workflow reduces the demand for solvents and energy-intensive recrystallization steps, contributing to a greener and more cost-effective manufacturing footprint. These efficiencies collectively enhance the competitiveness of the supply chain, allowing for more aggressive pricing strategies without compromising on margin.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts and the reduction in solvent usage for recrystallization lead to significant operational expenditure savings. By avoiding the need for expensive chiral resolution techniques or multiple purification passes, the overall processing time is drastically shortened, which lowers utility and labor costs per batch. The high yield means that less starting material is required to produce the same amount of final API, providing a buffer against fluctuations in raw material pricing. This economic efficiency is essential for maintaining profitability in the highly regulated and cost-sensitive pharmaceutical sector.
- Enhanced Supply Chain Reliability: The robustness of this chemical process ensures consistent batch-to-batch quality, which is a critical factor for reducing lead time for high-purity pharmaceutical intermediates. Because the reaction relies on common reagents like hydrochloric acid and thionyl chloride rather than specialized or scarce catalysts, the risk of supply disruption is minimized. The ability to scale the reaction from laboratory to commercial tonnage without re-optimizing the core chemistry provides confidence in long-term supply continuity. This reliability allows downstream manufacturers to plan their production schedules with greater certainty, mitigating the risks associated with inventory shortages.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, as demonstrated by successful trials ranging from gram to multi-kilogram scales without loss of efficiency. The reduction in waste generation aligns with increasingly strict environmental regulations, reducing the burden of waste treatment and disposal. By minimizing the release of hazardous by-products and optimizing atom economy, the manufacturer demonstrates a commitment to sustainable practices. This environmental stewardship not only reduces compliance costs but also enhances the brand reputation of the supply chain partners involved in the production of these critical antitumor medications.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this synthesis technology. These answers are derived directly from the experimental data and claims presented in the patent documentation to ensure accuracy and relevance for industry stakeholders. Understanding these details helps potential partners evaluate the feasibility of integrating this intermediate into their own manufacturing pipelines. It clarifies the operational requirements and the expected performance metrics associated with this advanced chemical process.
Q: How does this method improve Z-isomer selectivity compared to traditional routes?
A: Traditional methods often result in a Z/E ratio of roughly 2:1 or even favor the E-isomer under certain acidic conditions. This patented approach utilizes the solubility difference between Z and E isomers in concentrated hydrochloric acid. By crystallizing the Z-type hydrochloride salt out of the solution, the chemical equilibrium is continuously shifted, converting the remaining E-isomer into the desired Z-form, thereby achieving yields exceeding 85%.
Q: What are the critical reaction conditions for successful configuration conversion?
A: The concentration of hydrochloric acid is critical and must be maintained above 30%, with a volume at least 2 to 5 times the weight of the triaryl butyleneglycol substrate. Temperature control is also vital; while higher temperatures accelerate conversion, lower temperatures (around 10°C to 45°C) favor the crystallization of the Z-isomer hydrochloride, which is the driving force for the stereoselectivity.
Q: Is this process scalable for commercial production of antitumor intermediates?
A: Yes, the process is designed for scalability. The patent examples demonstrate successful scale-up from gram-scale laboratory experiments to 30kg batch sizes without loss of efficiency. The elimination of complex recrystallization steps and the use of common reagents like concentrated hydrochloric acid and thionyl chloride make it highly suitable for large-scale commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Toremifene Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of life-saving antitumor therapies. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent and reliable supply of materials. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of Toremifene intermediate meets the highest international standards. We understand that the complexity of stereoselective synthesis requires a partner with deep technical expertise, and our team is dedicated to navigating these challenges to deliver superior results for your projects.
We invite you to collaborate with us to leverage this advanced technology for your pharmaceutical development needs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our capabilities can enhance your supply chain efficiency. Let us be your trusted partner in bringing high-quality oncology medications to the market faster and more economically.