Scalable Production of Ketone Body Precursors via Novel Two-Step Esterification Technology
Scalable Production of Ketone Body Precursors via Novel Two-Step Esterification Technology
The metabolic potential of ketone bodies, specifically beta-hydroxybutyrate, has garnered immense attention in the pharmaceutical and nutritional sectors for treating neurological disorders, diabetes, and cardiovascular diseases. However, the direct administration of ketone bodies often leads to gastrointestinal distress and acidosis. To address these bioavailability and safety challenges, the industry requires efficient synthesis methods for ketone body esters, which act as stable, slow-release precursors. Patent CN113045416A discloses a groundbreaking preparation method for (R)-3-hydroxybutyryl-(R)-3-hydroxybutyl ester, a critical intermediate that hydrolyzes in vivo to release beneficial ketone bodies without the associated side effects of free acids or inorganic salts. This technology represents a paradigm shift from complex, multi-step protection strategies to a streamlined, industrially viable two-step process.
This report provides a deep technical and commercial analysis of this novel synthetic route, highlighting its superiority over prior art methods described in patents like WO2010021766 and WO2018226732. By leveraging simple tosylation and nucleophilic substitution chemistry, manufacturers can achieve total yields exceeding 70% with product purity greater than 95%, all while bypassing the need for expensive chromatographic purification. For R&D directors and procurement managers seeking a reliable pharmaceutical intermediate supplier, understanding the mechanistic robustness and cost-efficiency of this pathway is essential for securing a competitive supply chain in the growing metabolic health market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
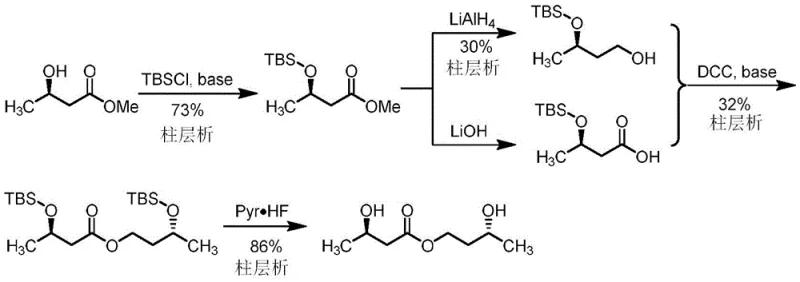
Prior to this innovation, the synthesis of (R)-3-hydroxybutyryl-(R)-3-hydroxybutyl ester was plagued by significant technical hurdles that hindered commercial scalability. As illustrated in the prior art route from Patent WO2018226732, traditional chemical synthesis relied heavily on protecting group chemistry, specifically using tert-butyl dimethyl silyl (TBS) groups to mask hydroxyl functionalities. This approach necessitates a lengthy sequence involving protection, reduction, condensation, and subsequent deprotection. The reliance on reagents like lithium aluminum hydride (LiAlH4) and coupling agents such as DCC introduces substantial safety risks and generates difficult-to-remove byproducts. Furthermore, the purification of intermediates in these conventional routes typically requires multiple rounds of column chromatography—often up to three times—which is economically prohibitive for large-scale manufacturing due to high solvent consumption and low throughput.
Enzymatic routes, such as those disclosed in WO2010021766, present a different set of challenges. While biocatalysis is often touted for its specificity, the alcoholysis of ethyl (R)-3-hydroxybutyrate with (R)-1,3-butanediol under reduced pressure creates a reaction mixture with extremely high viscosity. This physical property severely limits mass transfer efficiency, requiring extended reaction times of approximately three days and specialized equipment like rotary evaporators for the reaction itself. The downstream processing is equally cumbersome, involving filtration to remove enzymes followed by complex distillation steps to separate excess starting materials. These factors collectively result in poor process economics and difficulty in scaling up to meet the demands of a reliable pharmaceutical intermediate supplier.
The Novel Approach
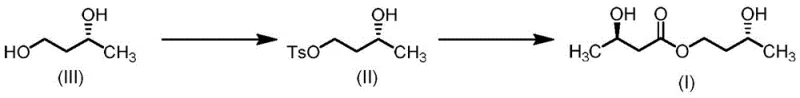
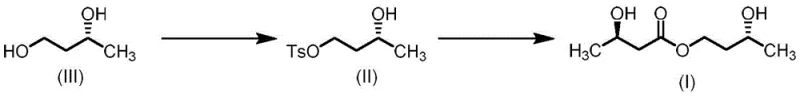
In stark contrast, the method disclosed in CN113045416A offers a remarkably concise and efficient alternative that eliminates the need for protecting groups entirely. The core innovation lies in the selective activation of (R)-1,3-butanediol (Compound III) via tosylation to form the monotosylate intermediate (Compound II), followed by a direct nucleophilic displacement with an (R)-3-hydroxybutyrate salt. This strategy capitalizes on the difference in reactivity between the primary and secondary hydroxyl groups of the diol, allowing for high regioselectivity without the need for temporary masking. The entire synthesis is accomplished in just two steps from readily available raw materials, drastically reducing the operational footprint and time-to-market for the final active ingredient.
The operational simplicity of this novel approach cannot be overstated. By avoiding the use of hazardous reducing agents and expensive coupling reagents, the process significantly lowers the barrier to entry for commercial production. The reaction conditions are mild, typically operating between 0°C and 90°C depending on the step, and utilize common organic solvents such as dichloromethane, toluene, or acetonitrile. Most importantly, the final product can be isolated through simple filtration and concentration, achieving a purity of over 95% without the need for column chromatography. This leap in process efficiency directly translates to cost reduction in API manufacturing, making the production of ketone body esters economically feasible for the first time on an industrial scale.
Mechanistic Insights into Tosylation and Nucleophilic Substitution
The success of this synthetic route hinges on the precise control of regioselectivity during the initial tosylation step. When (R)-1,3-butanediol reacts with p-toluenesulfonyl chloride in the presence of a base, the primary hydroxyl group at the 1-position is significantly more nucleophilic and less sterically hindered than the secondary hydroxyl group at the 3-position. By carefully controlling the stoichiometry of the base and the sulfonyl chloride—typically maintaining a molar ratio of roughly 1:1 to 1.2:1 relative to the diol—the reaction selectively generates the monotosylate (Compound II). Screening data indicates that solvent choice plays a critical role here; biphasic systems like dichloromethane/water or toluene/water often yield higher purity products compared to single-phase organic solvents, likely due to the differential solubility of the ionic byproducts and the suppression of bis-tosylation side reactions.
Following the formation of the tosylate, the second step involves an SN2-type nucleophilic substitution where the carboxylate anion of the (R)-3-hydroxybutyrate salt attacks the primary carbon bearing the tosyl group. The use of phase transfer catalysts, such as tetrabutylammonium bromide or iodide, is instrumental in facilitating this reaction, especially in less polar solvents like toluene. These catalysts enhance the solubility of the ionic hydroxybutyrate salt in the organic phase, thereby increasing the collision frequency between the nucleophile and the electrophilic tosylate. Crucially, because the reaction occurs at the primary carbon far removed from the chiral center at the 3-position of the butanediol backbone, the stereochemical integrity of the (R)-configuration is perfectly preserved. Similarly, the use of pre-formed (R)-3-hydroxybutyrate salts ensures that the chirality of the acid moiety is maintained, resulting in the desired (R,R)-diastereomer of the final ester with high optical purity.
How to Synthesize (R)-3-Hydroxybutyryl-(R)-3-Hydroxybutyl Ester Efficiently
Implementing this synthesis requires strict adherence to the optimized parameters regarding temperature, stoichiometry, and solvent systems to maximize yield and minimize impurities. The process begins with the activation of the diol, where maintaining low temperatures (0-5°C) during the addition of p-toluenesulfonyl chloride is vital to prevent exothermic runaway and side reactions. Following the isolation of the tosylate intermediate, the subsequent esterification is driven by heating the reaction mixture to reflux temperatures (75-90°C) for an extended period (12-24 hours) to ensure complete conversion. The detailed standardized synthesis steps, including specific workup procedures and quality control checkpoints, are outlined below for technical teams preparing for pilot scale-up.
- React (R)-1,3-butanediol (Compound III) with p-toluenesulfonyl chloride in a solvent like dichloromethane or toluene/water mixture using a base such as triethylamine or sodium carbonate to form the tosylate intermediate (Compound II).
- React the isolated Compound II with a salt of (R)-3-hydroxybutyric acid (e.g., sodium or potassium salt) in a solvent like toluene or acetonitrile, optionally using a phase transfer catalyst.
- Heat the reaction mixture to 75-90°C for 12-24 hours, then perform workup via filtration and concentration to obtain the final ester with high purity without column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition from enzymatic or protected-group synthesis to this direct tosylation-substitution route offers profound strategic benefits. The elimination of column chromatography is perhaps the most significant value driver, as chromatographic purification is notoriously resource-intensive, consuming vast quantities of silica gel and solvents while limiting batch sizes. By replacing this with simple crystallization or filtration, the new method drastically simplifies the manufacturing workflow, reduces solvent waste, and shortens the overall production cycle time. This efficiency gain allows for a substantial reduction in the cost of goods sold (COGS), enabling more competitive pricing for downstream pharmaceutical formulations.
- Cost Reduction in Manufacturing: The economic advantages of this process are derived from the use of commodity chemicals rather than specialized reagents. Replacing expensive coupling agents like DCC and protecting groups like TBS-Cl with inexpensive p-toluenesulfonyl chloride and inorganic bases significantly lowers raw material costs. Furthermore, the avoidance of cryogenic conditions (except for the initial addition) and vacuum distillation reduces energy consumption. The high yield and purity achieved without complex purification mean that less starting material is wasted, and the throughput per reactor volume is maximized, leading to significant cost optimization in pharmaceutical intermediate manufacturing.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the reliance on widely available, non-proprietary starting materials. (R)-1,3-butanediol and (R)-3-hydroxybutyric acid are commercially accessible in bulk quantities, reducing the risk of supply bottlenecks associated with niche enzymes or custom-synthesized protected intermediates. The robustness of the chemical process also means that production is less susceptible to the variability often seen in biocatalytic processes, such as enzyme batch-to-batch inconsistency or sensitivity to trace impurities. This stability ensures a consistent and reliable supply of high-purity intermediates for global clients.
- Scalability and Environmental Compliance: From an environmental and safety perspective, this route aligns well with green chemistry principles. The reduction in solvent usage and the elimination of heavy metal catalysts or hazardous reducing agents simplify waste treatment and disposal. The process does not require special equipment beyond standard glass-lined or stainless steel reactors, making it easy to scale from kilogram laboratory batches to multi-ton commercial production. This scalability ensures that as market demand for ketone body therapeutics grows, the supply infrastructure can expand seamlessly without requiring massive capital expenditure on new specialized facilities.
Frequently Asked Questions (FAQ)
To assist technical decision-makers in evaluating this technology for their specific applications, we have compiled answers to common questions regarding the process parameters, impurity profiles, and regulatory considerations. These insights are derived directly from the experimental data and embodiments provided in the patent literature, ensuring that the information is grounded in verified scientific results. Understanding these nuances is critical for integrating this intermediate into your existing development pipelines.
Q: What are the primary advantages of this synthesis method over enzymatic routes?
A: Unlike enzymatic alcoholysis which requires vacuum distillation and suffers from high viscosity and poor mass transfer, this chemical method operates under mild conditions with easily removable byproducts, eliminating the need for complex purification like column chromatography.
Q: How is stereochemical integrity maintained during the tosylation step?
A: The tosylation of the primary hydroxyl group in (R)-1,3-butanediol occurs without affecting the chiral center at the 3-position, ensuring that the (R)-configuration is preserved throughout the formation of the tosylate intermediate.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process uses cheap, commercially available reagents like p-toluenesulfonyl chloride and avoids specialized equipment or hazardous protection groups, making it highly scalable from kilogram to multi-ton production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (R)-3-Hydroxybutyryl-(R)-3-Hydroxybutyl Ester Supplier
The technological breakthroughs detailed in patent CN113045416A underscore the immense potential of (R)-3-hydroxybutyryl-(R)-3-hydroxybutyl ester as a key building block for next-generation metabolic therapeutics. At NINGBO INNO PHARMCHEM, we pride ourselves on being at the forefront of translating such innovative academic and patent research into commercial reality. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific solvent systems and reaction conditions required for this synthesis, ensuring that every batch meets stringent purity specifications through our rigorous QC labs and advanced analytical capabilities.
We invite pharmaceutical companies and research institutions to collaborate with us to accelerate the development of ketone body-based treatments. Our team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this streamlined synthesis can optimize your budget. Please contact our technical procurement team today to request specific COA data, route feasibility assessments, and samples to verify the quality of our intermediates. Let us help you secure a sustainable and cost-effective supply chain for your critical drug development projects.