Advanced Synthesis of Trelagliptin Degradation Impurity for High-Purity API Quality Control
Advanced Synthesis of Trelagliptin Degradation Impurity for High-Purity API Quality Control
In the highly regulated landscape of modern pharmaceutical manufacturing, the integrity of Active Pharmaceutical Ingredients (APIs) is paramount, particularly for chronic medications like Trelagliptin, a once-weekly DPP-4 inhibitor used for Type 2 diabetes. The patent CN114292240A, published in April 2022, introduces a pivotal advancement in the preparation of specific Trelagliptin degradation impurities, addressing a critical gap in quality control infrastructure. As global regulatory bodies such as the FDA and EMA enforce increasingly stringent limits on unidentified impurities—often mandating thresholds below 0.1% by weight—the ability to synthetically produce these degradation products for reference standards becomes a strategic necessity rather than a mere option. This technical insight report analyzes the patented methodology, which employs a rational two-step sequence involving nucleophilic substitution and subsequent hydrolysis, offering a robust pathway for generating high-purity reference materials essential for validating the safety profile of Trelagliptin formulations.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the acquisition of specific degradation impurities for pharmaceutical analysis has been fraught with significant technical and logistical challenges that hinder efficient quality assurance protocols. The conventional approach often relies on the forced degradation of the API itself under extreme stress conditions, such as high heat, intense UV radiation, or harsh acidic environments, followed by the arduous task of isolating trace impurities from the complex reaction matrix. This isolation process is inherently inefficient, typically yielding minute quantities of the target impurity mixed with numerous side products, necessitating resource-intensive purification techniques like preparative HPLC which drastically inflate costs and extend lead times. Furthermore, relying on degradation batches introduces variability; the impurity profile may shift unpredictably based on subtle changes in stress conditions, making it difficult to secure a consistent, certified reference standard required for rigorous batch-to-batch comparison and stability testing. The inability to reliably source these specific molecular structures creates a bottleneck in the release of finished drug products, as manufacturers cannot fully validate their analytical methods without authentic impurity standards.
The Novel Approach
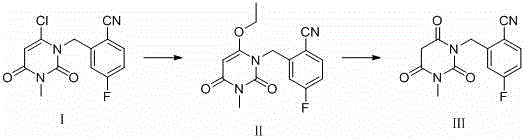
The methodology disclosed in patent CN114292240A represents a paradigm shift by moving from isolation to directed synthesis, utilizing a specifically designed precursor to construct the target impurity with high precision and yield. Instead of attempting to fish out trace molecules from a degraded soup, this novel approach starts with 2-[(6-chloro-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidyl)methyl]-4-fluorobenzonitrile (Compound I), a structurally defined intermediate that serves as a robust building block. By executing a controlled nucleophilic substitution followed by a targeted hydrolysis, the process constructs the degradation impurity (Compound III) directly, bypassing the chaotic mixture of byproducts associated with random degradation. This synthetic route is characterized by its strong operability and convenient purification; the final product precipitates out of the reaction mixture, allowing for simple filtration rather than complex chromatography. This transition from extraction to synthesis not only ensures a higher degree of structural certainty but also dramatically enhances the scalability of the process, enabling the production of gram-to-kilogram quantities of reference standards needed for comprehensive pharmaceutical development and regulatory submission.
Mechanistic Insights into Nucleophilic Substitution and Hydrolysis
The chemical elegance of this patented process lies in its two distinct mechanistic phases, each optimized to transform the starting material into the target degradation impurity with minimal side reactions. The first phase involves a nucleophilic substitution where the chlorine atom at the 6-position of the pyrimidine ring in Compound I is displaced by an ethoxy group from the ethanol solvent, facilitated by the presence of a mild base such as sodium bicarbonate. This transformation yields Compound II, an ethoxy-intermediate, which serves as a crucial pivot point in the synthesis; the introduction of the ethoxy group activates the molecule for the subsequent hydrolytic cleavage while maintaining the integrity of the sensitive benzonitrile and fluorophenyl moieties. The reaction conditions are carefully balanced, utilizing a mixture of ethanol and water at elevated temperatures (around 80°C) to drive the equilibrium towards the substitution product without inducing premature hydrolysis of the nitrile group or the uracil ring system, demonstrating a sophisticated understanding of chemoselectivity.
The second phase is an acid-catalyzed hydrolysis that converts the ethoxy-intermediate (Compound II) into the final carbonyl-containing impurity (Compound III), effectively mimicking the oxidative or hydrolytic degradation pathways that might occur during the shelf-life of the drug. By treating Compound II with an organic hydrochloric acid solution—options include hydrochloric acid in ethyl acetate, methanol, or dioxane—the ethoxy group is cleaved and converted into a carbonyl functionality, restoring the 2,4-dioxo structure characteristic of the degradation product. This step is remarkably versatile regarding temperature, proceeding efficiently between 10°C and 50°C over a period of 12 to 48 hours, which allows process engineers to tune the reaction rate based on thermal constraints. The mechanism likely proceeds through protonation of the ether oxygen, followed by nucleophilic attack by water and elimination of ethanol, a pathway that is clean and generates volatile byproducts that are easily removed, thereby simplifying the downstream purification and ensuring the final solid product meets the rigorous purity specifications demanded by pharmacopeial standards.
How to Synthesize Trelagliptin Impurity Efficiently
The operational protocol derived from this patent offers a streamlined workflow that minimizes unit operations while maximizing yield and purity, making it an ideal candidate for rapid implementation in quality control laboratories. The process begins with the dissolution of the chloro-precursor in a hydro-alcoholic medium, followed by a thermal treatment that drives the initial substitution to completion, after which the intermediate can be isolated or telescoped directly into the second step depending on the desired throughput. The subsequent acid treatment induces the final structural rearrangement, resulting in the precipitation of the target impurity as a solid filter cake, which can be washed with common organic solvents to remove residual salts and starting materials. This straightforward sequence eliminates the need for specialized equipment or hazardous reagents, relying instead on commodity chemicals and standard glassware, which significantly lowers the barrier to entry for manufacturing these critical reference standards. For a detailed breakdown of the specific molar ratios, solvent volumes, and precise thermal profiles required to replicate this synthesis with optimal results, please refer to the standardized guide below.
- Dissolve Compound I (2-[(6-chloro-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidyl)methyl]-4-fluorobenzonitrile) in a mixture of ethanol and water, add sodium bicarbonate as a base, and heat at 80°C for 12-24 hours to obtain Compound II.
- Treat Compound II with an organic hydrochloric acid solution (such as hydrochloric acid in ethyl acetate, methanol, or dioxane) at temperatures between 10°C and 50°C for 12-48 hours.
- Filter the reaction mixture to collect the precipitate, wash the filter cake with a suitable organic solvent like ethyl acetate or methanol, and dry to obtain the target Trelagliptin impurity (Compound III).
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this synthetic route offers substantial strategic benefits that extend far beyond the laboratory bench, directly impacting the cost structure and reliability of the pharmaceutical supply network. By shifting the sourcing model from extracting trace impurities to synthesizing them from abundant precursors, manufacturers can decouple their reference standard supply from the volatility of API degradation batches, ensuring a consistent and predictable inventory of critical quality control materials. The reliance on commercially available starting materials like the chloro-pyrimidine derivative and common solvents such as ethanol and ethyl acetate mitigates the risk of supply chain disruptions associated with exotic or proprietary reagents, fostering a more resilient procurement strategy. Furthermore, the simplicity of the workup procedure, which relies on filtration rather than energy-intensive distillation or chromatography, translates into significantly reduced processing times and lower utility consumption, driving down the overall cost of goods sold for these high-value specialty chemicals.
- Cost Reduction in Manufacturing: The economic viability of this process is underpinned by the elimination of expensive purification technologies; because the target impurity precipitates directly from the reaction mixture, the need for costly preparative chromatography columns and large volumes of HPLC-grade solvents is completely obviated. This reduction in downstream processing complexity leads to a drastic simplification of the manufacturing workflow, lowering labor costs and minimizing the loss of material that typically occurs during multiple purification cycles. Additionally, the use of sodium bicarbonate as a base and hydrochloric acid for hydrolysis utilizes some of the most cost-effective reagents available in the chemical industry, ensuring that the raw material costs remain negligible compared to the high value of the final certified reference standard. The cumulative effect of these efficiencies is a substantial optimization of the production budget, allowing for the allocation of resources towards other critical areas of pharmaceutical development without compromising on the quality or availability of essential impurity standards.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions provides a significant buffer against operational variability, ensuring that production schedules can be met with high confidence regardless of minor fluctuations in environmental parameters. The wide temperature tolerance for the hydrolysis step (10-50°C) means that the process does not require precise cryogenic cooling or high-energy heating, reducing the dependency on specialized HVAC systems and making the synthesis feasible in a broader range of manufacturing facilities. Moreover, the flexibility in solvent selection for the acid treatment—allowing for the use of ethyl acetate, methanol, or dioxane—empowers supply chain managers to adapt to local solvent availability or price fluctuations without needing to revalidate the entire process. This adaptability ensures continuous production capability, preventing delays in the release of Trelagliptin API batches due to a lack of qualified reference standards, thereby safeguarding the continuity of the drug supply for patients.
- Scalability and Environmental Compliance: The inherent design of this synthesis favors large-scale production, as the precipitation of the product simplifies the handling of reaction masses and avoids the bottlenecks associated with scaling chromatographic separations. From an environmental standpoint, the process generates minimal hazardous waste; the byproducts are primarily inorganic salts and volatile alcohols that can be easily managed or recycled, aligning with the principles of green chemistry and reducing the burden on waste treatment facilities. The avoidance of heavy metal catalysts or toxic oxidants further enhances the environmental profile of the process, facilitating easier regulatory compliance and reducing the costs associated with environmental health and safety (EHS) monitoring. This scalability ensures that as the demand for Trelagliptin grows globally, the supply of its corresponding impurity standards can be expanded seamlessly to match, supporting the long-term commercial viability of the drug product.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method, providing clarity for stakeholders evaluating its integration into their quality control workflows. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, ensuring that the information provided is grounded in verified scientific evidence. Understanding these nuances is critical for R&D directors and procurement officers who need to assess the feasibility of adopting this new standard for their internal testing protocols. By clarifying the operational parameters and the strategic advantages of this route, we aim to facilitate a smoother transition towards more robust and reliable impurity management strategies.
Q: Why is the synthesis of Trelagliptin degradation impurities critical for API manufacturing?
A: Regulatory agencies mandate strict limits (typically below 0.1%) for unidentified or toxic impurities in APIs. Synthesizing specific degradation impurities allows manufacturers to establish accurate analytical methods and quality control standards, ensuring the safety and efficacy of the final drug product.
Q: What are the key advantages of the patented two-step synthesis route?
A: The method utilizes readily available starting materials and avoids complex purification techniques. The process features strong operability, simple post-processing involving filtration, and effectively shortens the preparation cycle compared to isolation from degraded batches.
Q: Can this synthesis method be scaled for commercial reference standard production?
A: Yes, the process conditions are mild (10-80°C) and utilize common solvents like ethanol and ethyl acetate. The final product is obtained via filtration, which is highly scalable and avoids the bottlenecks associated with preparative chromatography, making it suitable for large-scale production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trelagliptin Impurity Supplier
At NINGBO INNO PHARMCHEM, we recognize that the synthesis of complex impurities like the Trelagliptin degradation product described in CN114292240A requires not just chemical expertise, but a deep commitment to quality and regulatory compliance. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need milligrams for method validation or kilograms for routine QC, we can deliver with consistency. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify the structural identity and purity of every batch, guaranteeing that our reference standards meet the exacting requirements of global pharmacopeias. Our dedication to technical excellence ensures that the impurities we supply serve as reliable anchors for your quality control systems, protecting the integrity of your final drug products.
We invite you to leverage our technical capabilities to optimize your supply chain for Trelagliptin and related DPP-4 inhibitor intermediates. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing prowess can support your regulatory filings and commercial production goals, ensuring a seamless partnership that drives value and reliability for your organization.