Advanced Preparation Technology for Everolimus Intermediates Enhancing Commercial Scalability
Advanced Preparation Technology for Everolimus Intermediates Enhancing Commercial Scalability
The pharmaceutical landscape for immunosuppressants and oncology treatments has been significantly shaped by the development of Everolimus, a potent derivative of Rapamycin. As demand for high-purity active pharmaceutical ingredients (APIs) intensifies, the efficiency of synthetic routes becomes a critical bottleneck for global supply chains. Patent CN103848849A introduces a transformative preparation technology that addresses long-standing inefficiencies in Everolimus manufacturing. This innovation focuses on a streamlined two-step process: the selective silylation of Rapamycin to form a key intermediate, followed by a mild deprotection sequence. By optimizing reaction conditions and reagent selection, this methodology achieves substantial improvements in both total yield and final product purity compared to legacy techniques. For R&D directors and procurement strategists, understanding these technical nuances is essential for securing a reliable everolimus intermediate supplier capable of meeting rigorous commercial standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
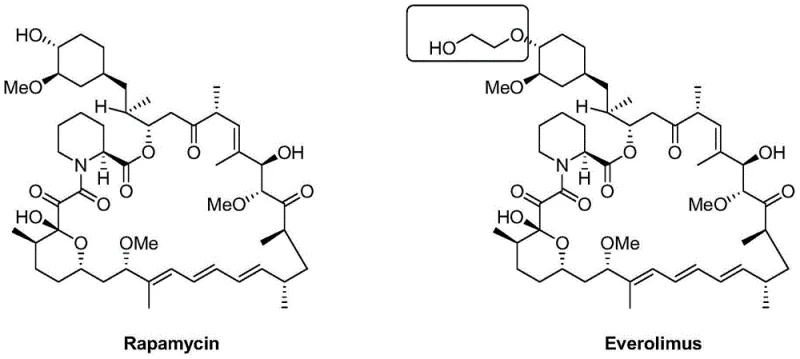
Historically, the synthesis of Everolimus has been plagued by low conversion rates and harsh reaction conditions that compromise scalability. Early methodologies, such as those described in US Patent 5665772, relied on the reaction of Rapamycin with trifluoromethanesulfonic acid 2-(tert-butyldimethylsilyloxy) ethyl ester in the presence of 2,6-lutidine. As illustrated in the reaction scheme below, this conventional Route 1 suffered from critically low yields, often hovering around merely 5%. The majority of the expensive Rapamycin raw material remained unconverted, and significant degradation of both reactants and products was observed. Furthermore, the subsequent deprotection step using 1N HCl in methanol frequently generated degraded impurities, resulting in low purity that necessitated complex and costly purification protocols. These inefficiencies render such methods economically unviable for large-scale industrial production.
The Novel Approach
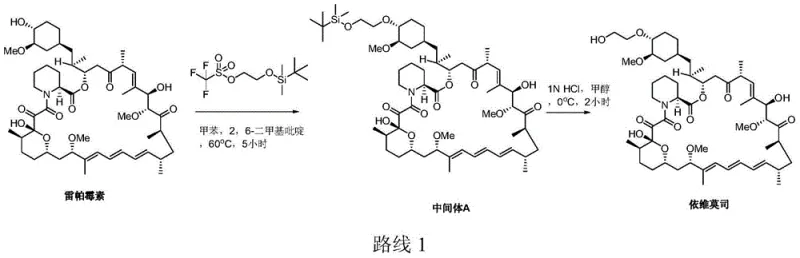
In stark contrast, the technology disclosed in CN103848849A represents a paradigm shift in process chemistry. The novel approach replaces the weak base 2,6-lutidine with sterically hindered, non-nucleophilic organic bases such as N,N-diisopropylethylamine (DIPEA). This strategic substitution dramatically enhances the nucleophilic attack on the silylating agent, driving the reaction towards completion with significantly higher conversion rates. Additionally, the process utilizes toluene as a preferred solvent and optimizes the molar ratios of reagents to minimize waste. The second step employs mild mineral acids like hydrochloric acid or sulfuric acid in water-miscible organic solvents such as acetone, avoiding the hazardous hydrogen fluoride used in other improved routes (like CN201010017955). This refinement not only boosts the yield of Intermediate A to over 60% but also ensures the final Everolimus crude product achieves a purity greater than 90%, streamlining the path to commercial viability.
Mechanistic Insights into DIPEA-Mediated Silylation and Acid Deprotection
The core of this technological advancement lies in the precise control of steric and electronic factors during the silylation of the 40-hydroxyl group on the Rapamycin macrocycle. Rapamycin possesses multiple hydroxyl groups, making regioselectivity a paramount challenge. The use of DIPEA, a bulky amine base, serves a dual purpose: it effectively scavenges the triflic acid byproduct without acting as a nucleophile itself, thereby preventing unwanted side reactions at other sensitive sites on the molecule. Unlike smaller bases that might coordinate too tightly or facilitate elimination reactions, DIPEA creates an environment where the 2-(tert-butyldimethylsilyloxy) ethyl trifluoromethanesulfonate selectively targets the most accessible and reactive 40-OH position. This selectivity is further enhanced by the choice of the tert-butyldimethylsilyl (TBDMS) protecting group, which strikes an optimal balance between stability during the reaction and lability during the workup.
Following the formation of Intermediate A, the deprotection mechanism is equally critical for maintaining product integrity. Traditional methods often resort to aggressive fluoride sources to cleave silyl ethers, which can induce skeletal rearrangements or decomposition of the sensitive macrolide ring. The patented process leverages the specific lability of the TBDMS group under mildly acidic conditions. By utilizing dilute hydrochloric acid (e.g., 0.5M) in acetone at controlled temperatures (0-35°C), the silicon-oxygen bond is cleaved efficiently to release the free hydroxyl group. This mild acidic environment preserves the delicate conjugated triene system and the ketone functionalities of the Everolimus structure, minimizing the formation of degradation byproducts. Consequently, the crude product emerging from this step requires minimal purification, directly translating to higher overall process efficiency and reduced solvent consumption.
How to Synthesize Everolimus Efficiently
The implementation of this optimized synthesis route requires strict adherence to the specified reaction parameters to maximize yield and purity. The process begins with the dissolution of Rapamycin and the organic base in an appropriate solvent, followed by the controlled addition of the silylating agent. Temperature control is vital, with the reaction typically maintained between 40°C and 70°C to ensure complete conversion without thermal degradation. Once Intermediate A is isolated and purified, the subsequent deprotection is conducted in a mixed solvent system compatible with aqueous acid. Detailed standard operating procedures regarding reagent grades, addition rates, and quenching protocols are essential for reproducibility. For a comprehensive guide on the specific experimental steps and workup procedures, please refer to the standardized synthesis instructions provided below.
- React Rapamycin with 2-(tert-butyldimethylsilyloxy) ethyl trifluoromethanesulfonate in toluene using DIPEA as a base at 60°C to form Intermediate A.
- Purify Intermediate A via column chromatography to remove unreacted starting materials and byproducts.
- Deprotect Intermediate A using 0.5M hydrochloric acid in acetone at 20°C to obtain Everolimus with greater than 90% crude purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this optimized synthesis technology offers profound strategic benefits beyond mere chemical elegance. The primary advantage lies in the drastic improvement of raw material utilization. By shifting from a process with single-digit yields to one demonstrating robust conversion, manufacturers can significantly reduce the cost of goods sold (COGS) associated with the expensive Rapamycin starting material. This efficiency gain mitigates the volatility of raw material pricing and reduces the inventory burden on the supply chain. Furthermore, the elimination of hazardous reagents like hydrogen fluoride simplifies the regulatory compliance landscape, reducing the overhead costs associated with specialized waste disposal and safety infrastructure. These factors collectively contribute to a more resilient and cost-effective supply chain for high-purity pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The adoption of DIPEA and mild acid deprotection eliminates the need for expensive and hazardous fluoride reagents, while the higher reaction yields mean less raw material is wasted per kilogram of finished product. This structural efficiency leads to substantial cost savings in the overall manufacturing budget without compromising quality.
- Enhanced Supply Chain Reliability: The use of common, commercially available solvents like toluene and acetone, along with stable reagents, ensures that production is not held hostage by the scarcity of exotic chemicals. This accessibility guarantees consistent production schedules and reduces the risk of delays caused by raw material shortages, ensuring a steady flow of intermediates to downstream API manufacturers.
- Scalability and Environmental Compliance: The mild reaction conditions (temperatures below 70°C and ambient pressure) make this process inherently safer and easier to scale from pilot plants to multi-ton commercial reactors. Additionally, the avoidance of heavy metal catalysts and toxic fluoride waste streams aligns with modern green chemistry principles, simplifying environmental permitting and reducing the ecological footprint of the manufacturing facility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Everolimus preparation technology. These insights are derived directly from the experimental data and comparative analysis presented in the patent literature. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this route for commercial production. The answers provided reflect the specific advantages of the optimized conditions over traditional methods.
Q: Why is DIPEA preferred over 2,6-lutidine in this Everolimus synthesis?
A: DIPEA (N,N-Diisopropylethylamine) offers superior steric hindrance and non-nucleophilic properties compared to 2,6-lutidine, significantly reducing side reactions and improving the conversion rate of Rapamycin to Intermediate A.
Q: What are the safety advantages of the deprotection step in Patent CN103848849A?
A: The process utilizes mild mineral acids like hydrochloric acid or sulfuric acid instead of hazardous hydrogen fluoride (HF) or pyridine complexes, drastically lowering operational risks and waste treatment costs.
Q: How does the TBDMS protecting group compare to TBDPS in this context?
A: The tert-butyldimethylsilyl (TBDMS) group used in this invention is less sterically bulky than tert-butyldiphenylsilyl (TBDPS), facilitating easier installation and, crucially, allowing for cleavage under much milder acidic conditions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Everolimus Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of complex molecules like Everolimus depends on mastering the intricate balance between chemical innovation and process engineering. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patents like CN103848849A are fully realized in practical manufacturing. We are committed to delivering stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of intermediate meets the exacting standards required by global regulatory bodies. Our capability to handle sensitive macrolide chemistry positions us as a strategic partner for pharmaceutical companies seeking to secure their supply chains against market fluctuations.
We invite you to collaborate with us to leverage these advanced synthesis technologies for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our optimized processes can drive value and reliability in your Everolimus supply chain.