Advanced Synthesis of 2-Chloro Trifluoromethyl Pyrimidine Intermediates for Pharmaceutical Manufacturing
Advanced Synthesis of 2-Chloro Trifluoromethyl Pyrimidine Intermediates for Pharmaceutical Manufacturing
The landscape of pharmaceutical and agrochemical intermediate manufacturing is constantly evolving, driven by the need for higher purity, better yields, and more sustainable processes. A pivotal advancement in this domain is detailed in patent CN102911123A, which discloses a robust preparation method for 2-chloro trifluoromethyl pyrimidine compounds. These heterocyclic structures are foundational building blocks in modern medicinal chemistry, particularly for developing anticancer agents, cardiovascular drugs, and antimicrobial therapies. The introduction of the trifluoromethyl group is strategically significant due to its strong electronegativity, high metabolic stability, and ability to enhance lipophilicity, thereby optimizing the pharmacokinetic profiles of drug candidates. This technical insight report analyzes the novel synthetic route described in the patent, highlighting its superiority over conventional methods and its implications for cost reduction in API manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art technologies, such as the method described in patent WO2010/134478A1, have historically faced significant bottlenecks that hinder efficient large-scale production. One major drawback is the reliance on single solvent systems, typically methanol, which restricts the reaction temperature to approximately 80°C. This thermal limitation is detrimental to reaction kinetics, preventing the system from reaching optimal conversion rates and ultimately capping the yield. Furthermore, the generation of alcohol as a byproduct in these single-solvent systems creates an equilibrium constraint; the accumulating alcohol inhibits the forward reaction, necessitating complex workup procedures or excessive reagent usage to drive completion. Additionally, the subsequent chlorination step in older methods often employs thionyl chloride with DMF as a solvent, a combination notorious for generating numerous byproducts and resulting in disappointingly low yields, which complicates downstream purification and increases waste disposal costs.
The Novel Approach
The methodology presented in CN102911123A offers a transformative solution to these entrenched processing challenges through a carefully engineered two-step synthesis. The innovation begins with the substitution of the single alcohol solvent with a mixed organic solvent system comprising a hydrocarbon, such as toluene, and an alcohol, such as ethanol. This strategic modification raises the reflux temperature ceiling to between 120°C and 130°C, dramatically accelerating the cyclization reaction. Crucially, the process incorporates a water trap (Dean-Stark apparatus) to continuously remove water and the alcohol byproduct generated during the reaction, effectively shifting the chemical equilibrium towards the desired 2-hydroxy trifluoromethyl pyrimidine intermediate. Following this, the chlorination step utilizes phosphorus oxychloride in acetonitrile with diisopropyl ethyl amine (DIPEA) as a catalyst, a refinement that boosts yield by more than 5% compared to non-catalyzed variants, ensuring a streamlined path to the final chloro-compound.
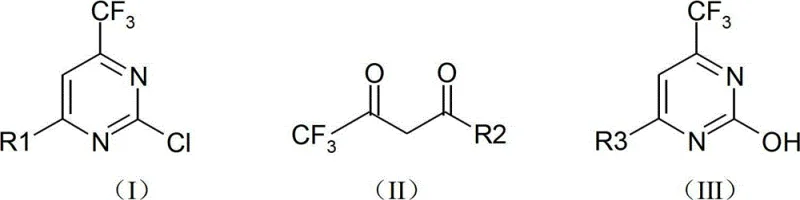
Mechanistic Insights into Base-Catalyzed Cyclization and Chlorination
The core of this synthesis lies in the base-catalyzed condensation and cyclization of urea with trifluoroacetyl derivatives. In the first stage, an alkaline catalyst, such as sodium ethylate, facilitates the nucleophilic attack of the urea nitrogen on the carbonyl carbon of the trifluoroacetyl derivative. The use of a mixed solvent system is not merely a physical change but a mechanistic enabler; the aromatic hydrocarbon component (toluene) forms an azeotrope with water and ethanol, allowing for the efficient physical removal of reaction byproducts via the water trap. This continuous removal prevents the reverse hydrolysis reaction, ensuring that the equilibrium heavily favors the formation of the cyclic pyrimidine ring. The reaction is maintained at 120-130°C for 8 to 16 hours, providing sufficient thermal energy to overcome activation barriers while the solvent system maintains a homogeneous phase conducive to molecular collision and bond formation.
In the second mechanistic phase, the transformation of the hydroxyl group to a chlorine atom is achieved through reaction with phosphorus oxychloride (POCl3). This step is critical for activating the pyrimidine ring for subsequent nucleophilic substitutions in drug synthesis. The inclusion of diisopropyl ethyl amine (DIPEA) serves a dual purpose: it acts as a proton scavenger to neutralize the HCl generated during the chlorination, and it likely coordinates with the phosphorus species to enhance the electrophilicity of the chlorinating agent. The reaction proceeds at elevated temperatures between 80°C and 150°C for 2 to 24 hours, ensuring complete conversion. Post-reaction, the mixture is treated with water under controlled low-temperature conditions (-2 to 4°C) to quench excess POCl3 safely, followed by alkalization to pH 8.5-10 to facilitate the extraction of the organic product, minimizing the retention of acidic impurities.
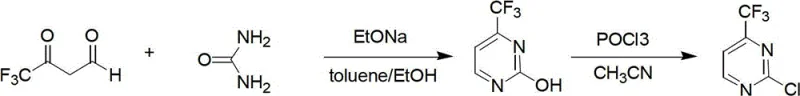
How to Synthesize 2-Chloro Trifluoromethyl Pyrimidine Efficiently
Implementing this synthesis route requires precise control over reaction parameters to maximize the reported yields of over 80%. The process begins with the charging of trifluoroacetyl derivatives, urea, and an alkaline catalyst into a reactor equipped with a water separator, using a toluene and ethanol mixture as the medium. The mixture is heated to reflux, and water is removed continuously until the reaction is complete, indicated by the cessation of water collection. After solvent removal and neutralization, the crude hydroxy-intermediate is isolated. This intermediate is then dissolved in acetonitrile, treated with phosphorus oxychloride, and heated before the dropwise addition of DIPEA. The detailed standardized operating procedures, including specific molar ratios and safety protocols for handling phosphorus oxychloride, are outlined in the structured guide below.
- Perform reflux reaction of urea and trifluoroacetyl derivatives using alkali catalyst in a toluene/ethanol mixture at 120-130°C, utilizing a water trap to remove moisture.
- Isolate the 2-hydroxy trifluoromethyl pyrimidine intermediate by removing solvents, neutralizing with acid, and extracting with organic solvent.
- React the hydroxy intermediate with phosphorus oxychloride and acetonitrile at 80-150°C using diisopropyl ethyl amine as a catalyst to obtain the final chloro compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented methodology represents a significant opportunity to optimize the cost structure and reliability of the supply chain for pyrimidine-based active pharmaceutical ingredients. By transitioning from low-yielding, single-solvent processes to this high-efficiency mixed-solvent system, manufacturers can drastically reduce the consumption of raw materials per kilogram of finished product. The elimination of problematic solvents like DMF in the chlorination step further simplifies waste management and reduces the environmental compliance burden, translating directly into lower operational expenditures. Moreover, the robustness of the reaction conditions, which tolerate a broader temperature range and utilize commodity solvents like toluene and ethanol, ensures that production schedules are less susceptible to disruptions caused by specialized reagent shortages or stringent storage requirements.
- Cost Reduction in Manufacturing: The primary economic driver of this technology is the substantial increase in overall yield, which exceeds 80% compared to the significantly lower yields of prior art. This efficiency gain means that for every ton of raw material purchased, a much higher volume of saleable intermediate is produced, effectively lowering the unit cost of goods sold. Additionally, the replacement of expensive or hazardous reagents like thionyl chloride/DMF systems with the POCl3/acetonitrile/DIPEA system reduces the cost of reagents and the associated costs of hazardous waste disposal. The simplified workup procedure, involving straightforward extraction and distillation rather than complex chromatographic purifications, further lowers labor and utility costs, delivering comprehensive cost reduction in API manufacturing.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of widely available, commodity-grade solvents such as toluene, ethanol, and acetonitrile, which are less prone to market volatility compared to specialized niche solvents. The process's tolerance for variation and its high conversion rate minimize the risk of batch failures, ensuring consistent output volumes that allow for more accurate forecasting and inventory planning. By reducing the dependency on complex, multi-step purification sequences that often act as bottlenecks in production, this method shortens the manufacturing cycle time, enabling faster response to market demand fluctuations and reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The design of this process is inherently scalable, moving seamlessly from laboratory bench scale to multi-ton commercial production without the need for exotic equipment. The use of a water trap to remove byproducts is a standard unit operation in chemical engineering, facilitating easy scale-up. From an environmental perspective, the avoidance of DMF and the efficient use of reagents result in a cleaner E-factor (mass of waste per mass of product). This aligns with increasingly stringent global environmental regulations, reducing the risk of regulatory shutdowns and positioning the supply chain as a sustainable partner for green chemistry initiatives in the pharmaceutical industry.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and benefits of this synthesis technology. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation, offering clarity on reaction mechanisms and process advantages. Understanding these details is crucial for R&D teams evaluating the feasibility of integrating this route into their existing manufacturing pipelines.
Q: Why is a mixed solvent system preferred over single solvents like methanol?
A: The use of a hydrocarbon and alcohol mixture, specifically toluene and ethanol, allows for higher reflux temperatures (120-130°C) compared to single alcohol solvents. Furthermore, the use of a water trap effectively removes water and generated alcohol during the reaction, shifting the equilibrium towards the product and significantly improving yield.
Q: What is the specific role of diisopropyl ethyl amine (DIPEA) in the chlorination step?
A: In the chlorination step using phosphorus oxychloride, diisopropyl ethyl amine acts as a crucial catalyst and acid scavenger. Its addition has been shown to improve the reaction yield by more than 5% compared to methods without it, ensuring a more efficient conversion of the hydroxy intermediate to the chloro derivative.
Q: How does this method compare to previous technologies regarding yield and industrial feasibility?
A: Unlike prior art which suffered from low yields and difficult solvent removal, this method achieves a total yield of over 80%. The process utilizes common industrial solvents and straightforward workup procedures (extraction and distillation), making it highly suitable for large-scale industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Chloro Trifluoromethyl Pyrimidine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of your drug development programs. As a leading CDMO and supplier, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from pilot plant to full-scale manufacturing is seamless and efficient. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 2-chloro trifluoromethyl pyrimidine compound adheres to the highest international standards. We are committed to leveraging advanced synthetic methodologies, such as the one described in CN102911123A, to deliver products that empower your research and commercialization efforts.
We invite you to collaborate with us to explore how this optimized synthesis route can benefit your specific project requirements. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your volume needs, demonstrating the tangible economic benefits of switching to this high-yield process. Please contact our technical procurement team today to request specific COA data for our current stock or to discuss route feasibility assessments for your custom synthesis projects, ensuring a secure and cost-effective supply chain for your future successes.