Scalable Synthesis of 6-Amino-5-Fluoro-1-Isoindolinone for Commercial Pharmaceutical Applications
The pharmaceutical industry constantly seeks robust synthetic routes for complex heterocyclic intermediates that serve as critical building blocks for next-generation therapeutics. Patent CN102911109A introduces a transformative preparation method for 6-amino-5-fluoro-1-isoindolinone, a valuable scaffold in medicinal chemistry known for its potential in oncology and antimicrobial applications. This technical disclosure addresses the longstanding inefficiencies associated with previous synthetic methodologies by proposing a streamlined four-step sequence that begins with the readily available starting material, 4-fluoro-2-methylbenzoic acid. By shifting away from exotic reagents and energy-intensive conditions, this innovation offers a pathway that is not only chemically elegant but also commercially viable for large-scale manufacturing. For R&D directors and process chemists, understanding the nuances of this route is essential for evaluating its integration into existing supply chains for high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this invention, the synthesis of 6-amino-5-fluoro-1-isoindolinone was predominantly governed by methods described in patents such as WO2008079759 and WO2008079836, which presented significant barriers to industrial adoption. These legacy routes typically involved a cumbersome five-step sequence that suffered from critically low overall yields, often calculating to merely 2.58% based on the initial starting material. A major bottleneck in these conventional processes was the reliance on highly toxic copper cyanide (CuCN) reagents during the fourth step, posing severe safety hazards and environmental compliance challenges for manufacturing facilities. Furthermore, these older methods frequently necessitated the use of microwave radiation to drive reactions, a condition that is notoriously difficult to scale up from laboratory glassware to multi-ton reactors. The combination of hazardous reagents, energy-intensive equipment requirements, and poor atom economy rendered these traditional approaches economically unfeasible for reliable agrochemical intermediate or pharmaceutical supplier operations seeking cost-effective production.
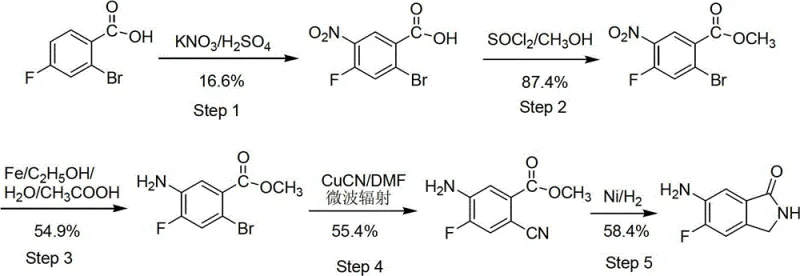
The Novel Approach
In stark contrast to the limitations of the prior art, the methodology outlined in CN102911109A utilizes a strategic four-step design that prioritizes operational simplicity and safety without compromising on yield. The process initiates with a straightforward esterification of 4-fluoro-2-methylbenzoic acid, followed by a tandem bromination and cyclization sequence that efficiently constructs the isoindolinone core. By replacing toxic cyanide sources with standard radical bromination agents like N-bromosuccinimide (NBS) and utilizing common solvents such as carbon tetrachloride and tetrahydrofuran, the new route drastically simplifies the hazard profile of the synthesis. The subsequent nitration and reduction steps are conducted under mild conditions that do not require specialized microwave equipment, allowing for seamless translation from pilot plant to commercial scale. This novel approach demonstrates how thoughtful reagent selection and step convergence can lead to substantial cost savings in electronic chemical manufacturing and broader fine chemical sectors by eliminating unnecessary complexity.
Mechanistic Insights into Regioselective Nitration and Cyclization
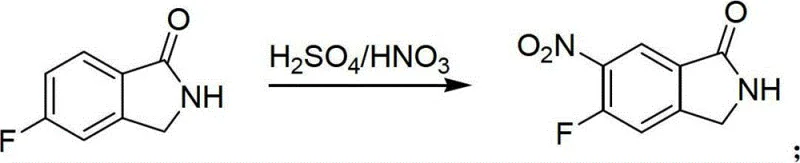
The success of this synthetic strategy hinges on the precise control of regioselectivity during the functionalization of the aromatic ring, particularly in the nitration step. When 5-fluoro-1-isoindolinone is subjected to mixed acid conditions comprising concentrated sulfuric acid and fuming nitric acid, the electrophilic aromatic substitution occurs selectively at the 6-position. This selectivity is driven by the electronic effects of the existing fluorine substituent and the lactam nitrogen, which direct the incoming nitronium ion to the position ortho to the fluorine and para to the nitrogen bridgehead. Maintaining strict temperature control, often utilizing ice bath cooling during the addition of fuming nitric acid, is critical to preventing over-nitration or oxidative degradation of the sensitive isoindolinone ring system. The mechanistic precision ensures that the resulting 6-nitro-5-fluoro-1-isoindolinone is formed with high purity, minimizing the formation of isomeric impurities that would be difficult to separate in downstream processing.
Furthermore, the cyclization mechanism employed in the second step represents a clever utilization of intramolecular nucleophilic attack to close the five-membered ring. Following the radical bromination of the methyl group to form the bromomethyl intermediate, the introduction of concentrated ammonia provides the nucleophile necessary to displace the bromide and simultaneously attack the ester carbonyl. This one-pot transformation effectively builds the heterocyclic core while installing the nitrogen atom required for the final structure. The avoidance of transition metal catalysts in this cyclization step is particularly noteworthy for supply chain stability, as it removes the risk of heavy metal contamination in the final API. For quality assurance teams, this means that the burden of proving residual metal limits is significantly reduced, streamlining the regulatory filing process for new drug applications that utilize this intermediate.
How to Synthesize 6-Amino-5-Fluoro-1-Isoindolinone Efficiently
Implementing this synthesis requires careful attention to stoichiometry and workup procedures to maximize the impressive 14.99% overall yield reported in the patent data. The process begins with the acid-catalyzed esterification of 4-fluoro-2-methylbenzoic acid in methanol, followed by a radical bromination using NBS and benzoyl peroxide initiator. The subsequent cyclization with ammonia and final nitration/reduction sequence demands precise control of reaction times and temperatures to ensure optimal conversion. While the chemical transformations are robust, the efficiency of the isolation steps—specifically the filtration and washing protocols—is paramount for achieving the high purity required for pharmaceutical use. The detailed standardized synthesis steps see the guide below for specific operational parameters.
- Esterify 4-fluoro-2-methylbenzoic acid with methanol and sulfuric acid to form the methyl ester intermediate.
- Perform radical bromination followed by cyclization with ammonia to generate the 5-fluoro-1-isoindolinone core.
- Execute regioselective nitration using fuming nitric acid and sulfuric acid to introduce the nitro group at the 6-position.
- Reduce the nitro group to an amine using Pd/C catalyst under hydrogen atmosphere to yield the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the shift from the legacy five-step route to this optimized four-step process represents a significant opportunity for cost reduction in pharmaceutical intermediate manufacturing. The elimination of column chromatography purification across all steps is a primary driver of economic efficiency, as chromatographic separation is often the most expensive and time-consuming unit operation in fine chemical production. By relying on simple crystallization, filtration, and solvent washing for isolation, the process drastically reduces solvent consumption and labor hours, leading to a leaner cost structure. Additionally, the use of commodity chemicals like methanol, sulfuric acid, and ammonia ensures that raw material sourcing remains stable and unaffected by the volatility often seen with specialized organometallic reagents. This stability translates directly into enhanced supply chain reliability, allowing manufacturers to maintain consistent inventory levels without the risk of disruption from niche reagent shortages.
- Cost Reduction in Manufacturing: The economic benefits of this route are derived principally from the simplification of the purification workflow and the avoidance of precious metal catalysts in the early stages. By eliminating the need for expensive silica gel columns and the associated solvent recovery systems, the operational expenditure per kilogram of product is significantly lowered. Furthermore, the removal of toxic copper cyanide negates the need for specialized waste treatment protocols required for cyanide disposal, which adds substantial hidden costs to the legacy process. The overall yield improvement from roughly 2.58% to nearly 15% means that less starting material is required to produce the same amount of final product, effectively multiplying the throughput of existing reactor capacity without capital investment.
- Enhanced Supply Chain Reliability: From a logistics perspective, the reliance on bulk commodity chemicals rather than exotic reagents ensures a resilient supply chain capable of withstanding market fluctuations. The starting material, 4-fluoro-2-methylbenzoic acid, is widely available from multiple global suppliers, reducing the risk of single-source dependency. The mild reaction conditions, which do not require high-pressure microwave reactors or cryogenic temperatures below -78°C, allow the synthesis to be performed in standard glass-lined or stainless steel reactors found in most contract manufacturing organizations. This compatibility with standard infrastructure reduces lead time for high-purity pharmaceutical intermediates by facilitating faster technology transfer between sites and enabling rapid scale-up to meet sudden increases in demand.
- Scalability and Environmental Compliance: The environmental footprint of this synthesis is markedly smaller than that of the prior art, aligning with modern green chemistry principles and increasingly strict regulatory standards. The absence of heavy metals like copper in the synthetic route simplifies the environmental impact assessment and reduces the burden on wastewater treatment facilities. The process generates fewer hazardous byproducts, and the solvents used, such as methanol and ethyl acetate, are easier to recover and recycle compared to the complex mixtures often generated in cyanide chemistry. This improved environmental profile not only lowers compliance costs but also enhances the sustainability credentials of the final drug product, a factor that is becoming increasingly important for pharmaceutical companies aiming to meet corporate social responsibility goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route, based on the specific data points and advantages highlighted in the patent literature. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this method for their own production lines. The answers provided reflect the empirical results observed during the optimization of the process, ensuring that expectations regarding yield and purity are grounded in experimental reality.
Q: What is the overall yield advantage of this new synthesis route compared to prior art?
A: The novel route described in CN102911109A achieves an overall yield of approximately 14.99%, which is a substantial improvement over the previously reported 2.58% yield found in older patents like WO2008079759.
Q: Does this process require toxic reagents like copper cyanide?
A: No, this optimized method eliminates the need for highly toxic copper cyanide (CuCN) and avoids harsh microwave radiation conditions, making it significantly safer and more suitable for large-scale industrial production.
Q: Is column chromatography required for purification in this method?
A: No, a key commercial advantage of this process is that intermediates and the final product can be isolated through simple crystallization, filtration, and washing, completely removing the need for expensive and time-consuming column chromatography.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-Amino-5-Fluoro-1-Isoindolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving medications, and we are committed to delivering excellence in every batch. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need material for clinical trials or full-scale commercial launch, we can meet your volume requirements with precision. We operate under stringent purity specifications and utilize rigorous QC labs equipped with advanced analytical instrumentation to guarantee that every shipment of 6-amino-5-fluoro-1-isoindolinone meets the highest international standards. Our dedication to quality assurance means that we proactively monitor impurity profiles and optimize crystallization processes to deliver a product that facilitates smooth downstream synthesis for our partners.
We invite you to collaborate with us to leverage the efficiencies of this advanced synthetic route for your specific project needs. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your annual volume requirements. We are ready to provide specific COA data from recent batches and conduct comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your drug development timeline. Let us be your partner in transforming complex chemical challenges into commercial successes.