Revolutionizing Carborane Functionalization: A Scalable Rh-Catalyzed B-H Activation Strategy for High-Purity Intermediates
The landscape of organoboron chemistry is undergoing a significant transformation, driven by the urgent need for safer and more efficient synthetic routes for boron-rich clusters. Patent CN112028919A introduces a groundbreaking B(3)/B(3,6)-o-carborane alkylation method that fundamentally shifts the paradigm from hazardous pre-functionalization to direct catalytic activation. This innovation addresses long-standing challenges in synthesizing carborane derivatives, which are critical components in Boron Neutron Capture Therapy (BNCT) and advanced luminescent materials. By utilizing a Rhodium(I) catalytic system coupled with N-acyl-diimide active amides, the technology enables the precise installation of organic fragments onto the electron-deficient boron cage without the need for toxic thallium salts or aggressive oxidizing agents. For R&D directors and procurement specialists, this represents a pivotal opportunity to secure a reliable pharmaceutical intermediate supplier capable of delivering high-purity building blocks through a streamlined, environmentally compliant process.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the selective functionalization of the B(3) position on o-carborane cages has been plagued by severe synthetic inefficiencies and safety hazards. Traditional strategies typically rely on pre-functionalized precursors, necessitating multi-step sequences that often involve the use of equivalent amounts of highly toxic thallium acetate (TlOAc) or dangerous reagents like liquid ammonia and metallic sodium. These legacy processes not only pose significant environmental risks due to heavy metal waste but also suffer from poor atom economy and low overall yields caused by cumulative losses across multiple reaction stages. Furthermore, the harsh reaction conditions required, such as sealed tube heating at 110°C in THF or the use of strong Lewis acids like BI3, severely limit the scope of compatible substrates, rendering many complex drug-like molecules inaccessible for carborane modification. This lack of functional group tolerance creates a bottleneck for medicinal chemists attempting to incorporate boron clusters into late-stage drug candidates.
The Novel Approach
In stark contrast, the methodology disclosed in CN112028919A leverages a sophisticated Rhodium-catalyzed cross-coupling strategy that bypasses the need for pre-activation entirely. By employing N-acyl-diimide active amides as robust coupling partners, the reaction proceeds through a direct B-H activation mechanism that is both oxidant-free and base-free. This eliminates the risk of cage degradation often associated with strong bases and prevents the formation of unwanted deboronation byproducts. The process operates under relatively mild thermal conditions using toluene as a solvent, which enhances the solubility of larger organic substrates compared to previous low-temperature protocols. Consequently, this approach offers superior functional group compatibility, tolerating sensitive moieties such as esters, ketones, and heterocycles, thereby expanding the chemical space available for designing next-generation carborane-based therapeutics and materials.
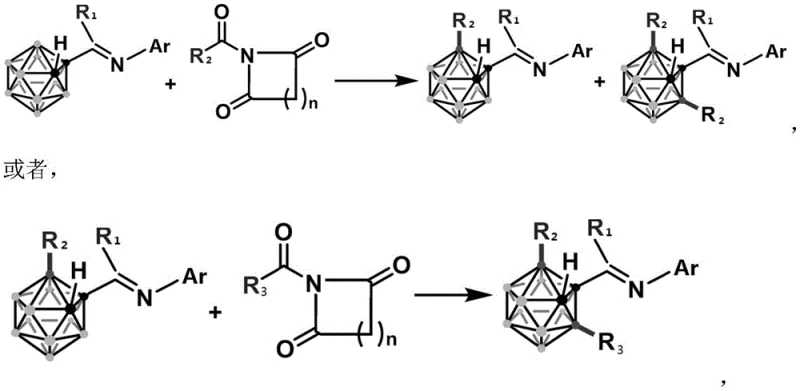
Mechanistic Insights into Rhodium-Catalyzed B-H Activation
The core of this technological advancement lies in the unique interaction between the Rhodium(I) catalyst and the N-acyl-diimide moiety, which facilitates a two-step oxidative addition into the B-H bond. Unlike electrophilic substitution methods that struggle with the electron-deficient nature of the B(3) site, this catalytic cycle effectively activates the inert boron-hydrogen bond through a concerted metalation-deprotonation pathway or similar oxidative mechanism. The use of an imine directing group on the carborane cage carbon plays a crucial role in guiding the regioselectivity towards the B(3) and B(3,6) positions, overcoming the inherent electronic bias of the cluster. This precise control ensures that the desired mono- and di-substituted products are formed with high selectivity, minimizing the formation of inseparable isomers that often complicate downstream purification efforts in traditional syntheses.
Furthermore, the generation of amine-based anions during the catalytic cycle negates the requirement for external strong bases, which is a critical factor in preserving the integrity of the carborane cage. In conventional nucleophilic substitutions, the presence of strong bases can trigger cage opening or deboronation, leading to complex mixtures and reduced yields. By avoiding these harsh conditions, the new method maintains the structural stability of the closo-carborane framework throughout the reaction. This mechanistic elegance translates directly to practical benefits, such as simplified workup procedures and higher isolated yields, making it an attractive option for the commercial scale-up of complex pharmaceutical intermediates where purity and reproducibility are paramount.
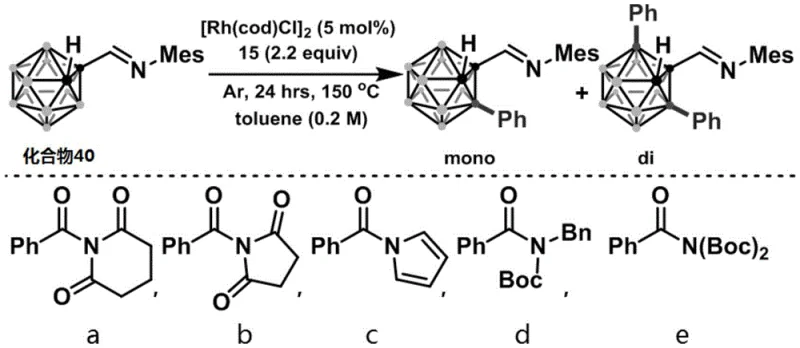
How to Synthesize B(3)/B(3,6)-o-Carborane Alkylated Compounds Efficiently
Implementing this synthesis route involves a straightforward protocol that begins with the preparation of the active amide coupling partner from readily available carboxylic acids. The subsequent cross-coupling reaction is performed in a standard Schlenk tube under an inert argon atmosphere, utilizing [Rh(cod)Cl]2 as the precatalyst and toluene as the solvent at 150°C. The detailed standardized synthesis steps see the guide below.
- Derivatize the target compound into an N-acyl-diimide active amide using standard coupling reagents or acid chloride methods.
- Combine the active amide with an imine-substituted o-carborane and a Rhodium catalyst such as [Rh(cod)Cl]2 in an inert atmosphere.
- Heat the reaction mixture in toluene at 150°C for 24 hours, then purify the mono- and di-substituted products via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this Rh-catalyzed alkylation method offers substantial strategic advantages beyond mere technical performance. The elimination of toxic thallium reagents and hazardous oxidants drastically simplifies waste management protocols and reduces the regulatory burden associated with handling dangerous chemicals. This shift towards greener chemistry not only aligns with global sustainability goals but also mitigates the risk of supply chain disruptions caused by strict environmental regulations on hazardous material transport and disposal. Additionally, the use of stable carboxylic acid-derived amides as starting materials ensures a consistent and reliable supply of raw materials, unlike specialized halogenated precursors that may have limited availability or long lead times.
- Cost Reduction in Manufacturing: The streamlined single-step coupling process significantly reduces operational costs by eliminating the need for multi-step pre-functionalization sequences. By removing expensive and toxic reagents like TlOAc and avoiding complex purification steps required to remove heavy metal residues, the overall cost of goods sold is substantially lowered. The high atom economy and improved yields further contribute to cost reduction in pharmaceutical intermediate manufacturing, allowing for more competitive pricing structures without compromising on quality standards.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions and the wide availability of starting materials enhance the reliability of the supply chain. The method's tolerance to various functional groups means that a single synthetic platform can be adapted for a diverse range of products, reducing the need for specialized equipment or distinct process lines for different intermediates. This flexibility ensures reducing lead time for high-purity carborane derivatives, enabling faster response to market demands and accelerating the timeline from discovery to clinical supply.
- Scalability and Environmental Compliance: Demonstrated success in gram-scale synthesis confirms the viability of this method for large-scale production. The use of common solvents like toluene and the absence of moisture-sensitive reagents facilitate easier scale-up in standard reactor systems. Moreover, the reduced environmental footprint supports compliance with increasingly stringent global environmental standards, positioning manufacturers as responsible partners in the global pharmaceutical supply chain.
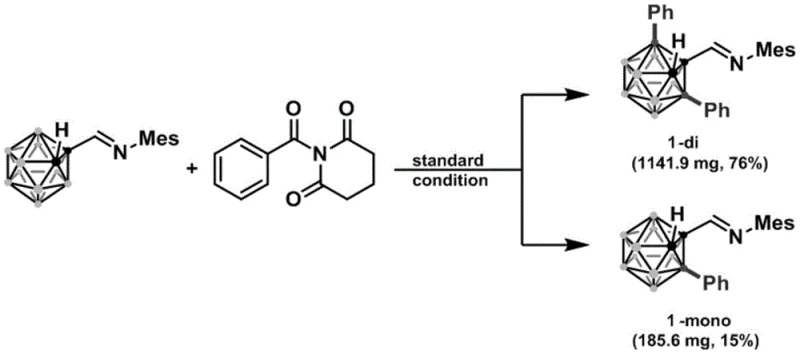
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel carborane alkylation technology. These insights are derived directly from the experimental data and comparative analysis presented in the patent documentation, providing clarity on its practical application and benefits for industrial partners.
Q: What are the advantages of this Rh-catalyzed method over traditional carborane functionalization?
A: Unlike traditional methods requiring toxic thallium reagents or harsh pre-functionalization steps, this protocol achieves direct B-H activation under oxidant-free conditions, significantly improving safety and functional group tolerance.
Q: Is this synthesis method scalable for industrial production?
A: Yes, the patent demonstrates successful gram-scale synthesis with high yields (up to 76% for di-substituted products), indicating strong potential for commercial scale-up without complex purification bottlenecks.
Q: What types of functional groups are compatible with this alkylation process?
A: The method exhibits excellent compatibility with diverse groups including esters, ketones, halides, and heterocycles, making it suitable for late-stage functionalization of complex drug molecules.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable o-Carborane Derivatives Supplier
As the demand for boron-containing therapeutics and advanced materials continues to surge, partnering with a technically proficient manufacturer is essential for success. NINGBO INNO PHARMCHEM stands at the forefront of this evolution, leveraging cutting-edge catalytic technologies to deliver superior carborane intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and efficiency. We operate stringent purity specifications and maintain rigorous QC labs to guarantee that every batch meets the highest international standards, providing you with the confidence needed for critical drug development programs.
We invite you to collaborate with us to explore the full potential of this innovative alkylation chemistry for your specific applications. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your project requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to help you optimize your supply chain and accelerate your time to market.