Advanced Synthesis of Carfilzomib Impurity V for Quality Control and Regulatory Compliance
The pharmaceutical industry faces rigorous challenges in maintaining the safety and efficacy of oncology treatments, particularly for complex proteasome inhibitors like Carfilzomib. As detailed in patent CN114262359A, the control of specific process-related impurities is critical for regulatory approval and patient safety. This patent discloses a novel, streamlined preparation method for Carfilzomib Impurity V, a critical reference standard required for quality control during the manufacturing of the active pharmaceutical ingredient (API). Unlike traditional methods that struggle with isolation, this technology offers a dedicated synthetic route that ensures high purity and yield without relying on complex purification techniques like preparative chromatography. For R&D directors and quality assurance teams, access to such well-characterized impurity standards is indispensable for validating analytical methods and establishing strict acceptance criteria for the final drug product.
Carfilzomib is a potent treatment for multiple myeloma, and its synthesis involves intricate peptide coupling and epoxidation steps. During these processes, residual starting materials can react further to generate structurally related byproducts. Specifically, the patent highlights how residual Compound I can undergo epoxidation and subsequent reactions to form Impurity V. The presence of such impurities, even in trace amounts, can impact the safety profile of the injection. Therefore, having a reliable supply of authentic Impurity V is not just a regulatory checkbox but a fundamental requirement for robust process development. The disclosed method addresses the scarcity of this specific reference material by providing a scalable, three-step synthesis that bypasses the bottlenecks of traditional isolation.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, obtaining specific impurities like Compound V for Carfilzomib has been a significant bottleneck in process chemistry. In conventional manufacturing scenarios, these impurities are generated incidentally in minute quantities alongside the main product. Isolating them typically requires labor-intensive and costly techniques such as column chromatography or preparative HPLC. These methods are inherently inefficient for large-scale production of reference standards due to low recovery rates and the consumption of vast amounts of silica gel and solvents. Furthermore, the instability of certain intermediates during prolonged chromatographic separation can lead to degradation, compromising the purity of the isolated impurity. This makes it difficult for quality control laboratories to obtain sufficient quantities of high-purity material needed for method validation and stability studies.
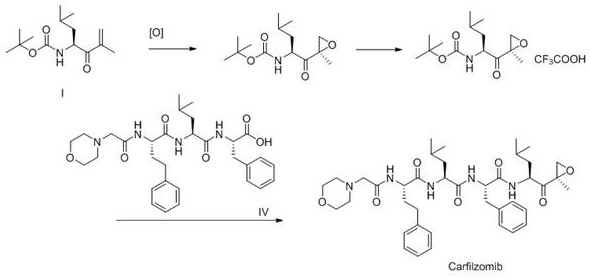
The Novel Approach
The technology described in CN114262359A revolutionizes this process by shifting from isolation to dedicated synthesis. Instead of fishing for needles in a haystack, the inventors have designed a linear pathway starting from a specific alkene precursor (Compound I). This approach allows for the deliberate construction of the impurity structure through controlled chemical transformations. By utilizing standard unit operations like catalytic hydrogenation and acid-mediated deprotection, the method achieves high conversion rates. The final product is purified via simple recrystallization rather than chromatography, drastically reducing solvent waste and processing time. This strategic shift not only guarantees a consistent supply of the impurity but also ensures that the material produced is chemically identical to the process-related impurity found in the API, making it a perfect match for analytical calibration.
Mechanistic Insights into the Three-Step Synthetic Route
The core of this innovation lies in a concise three-step sequence that transforms a protected amino acid derivative into the target impurity. The first step involves the catalytic hydrogenation of the olefinic double bond in Compound I. Using Palladium on Carbon (Pd/C) as a heterogeneous catalyst, the reaction proceeds under mild conditions (0-30°C) in solvents such as methanol or dichloromethane. This reduction is highly selective, saturating the carbon-carbon double bond without affecting other sensitive functional groups like the Boc-protected amine or the ketone moiety. The use of Pd/C is particularly advantageous for scale-up, as the catalyst can be easily removed by filtration, leaving a clean reaction mixture ready for the next step.
Following hydrogenation, the second step executes the removal of the tert-butoxycarbonyl (Boc) protecting group. This is achieved using Trifluoroacetic Acid (TFA) in dichloromethane at low temperatures ranging from -20°C to 0°C. The acidic conditions cleave the carbamate linkage, generating the free amine salt (Compound III) which is crucial for the subsequent coupling reaction. The final step is an amide bond formation between the newly liberated amine (Compound III) and the Carfilzomib main chain fragment (Compound IV). This peptide coupling is facilitated by modern condensing agents like HBTU, HOBT, or PyBOP in the presence of an organic base such as DIPEA. This activation strategy ensures rapid and efficient bond formation, minimizing racemization and side reactions, ultimately yielding the target Impurity V with exceptional purity.
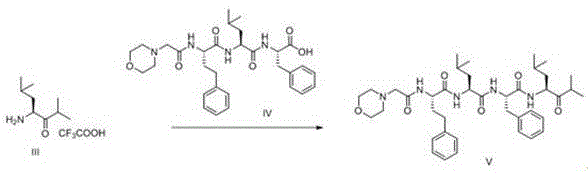
How to Synthesize Carfilzomib Impurity V Efficiently
The synthesis protocol outlined in the patent provides a robust framework for producing this critical reference standard. The process is designed to be operationally simple, avoiding the need for specialized equipment beyond standard reactor vessels capable of handling hydrogenation and low-temperature reactions. The key to success lies in the careful control of stoichiometry during the coupling phase and the selection of appropriate recrystallization solvents to maximize recovery. Below is the structural overview of the standardized synthesis procedure derived from the patent claims.
- Hydrogenate Compound I using Palladium on Carbon (Pd/C) catalyst in solvents like methanol or dichloromethane to reduce the double bond, yielding Compound II.
- Remove the Boc protecting group from Compound II using Trifluoroacetic Acid (TFA) in dichloromethane at low temperatures (-20 to 0°C) to obtain Compound III.
- Couple Compound III with the Carfilzomib main chain (Compound IV) using condensing agents such as HBTU or PyBOP and an organic base to form the final Impurity V.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits in terms of cost efficiency and supply reliability. The elimination of column chromatography is a major cost driver reduction, as it removes the need for expensive silica gel, large volumes of high-grade solvents, and the associated waste disposal costs. Additionally, the use of commodity chemicals like Pd/C, TFA, and common organic solvents ensures that raw material sourcing is straightforward and not subject to the volatility of specialized reagent markets. This stability in the supply chain is crucial for maintaining continuous production of quality control standards without interruption.
- Cost Reduction in Manufacturing: The process significantly lowers the cost of goods sold (COGS) for the impurity standard by replacing low-yield isolation techniques with high-yield synthetic steps. The ability to purify the final product via recrystallization instead of chromatography translates to substantial savings in both material and labor costs. Furthermore, the high atom economy of the hydrogenation and coupling steps ensures that raw materials are utilized efficiently, minimizing waste generation and enhancing the overall economic viability of the production process.
- Enhanced Supply Chain Reliability: By relying on widely available catalysts and reagents, the risk of supply disruption is minimized. The reaction conditions are mild and do not require extreme pressures or temperatures, which reduces the strain on manufacturing equipment and lowers maintenance costs. This operational simplicity allows for flexible production scheduling, enabling suppliers to respond quickly to fluctuating demand from pharmaceutical clients who require batch-specific impurity profiles for their regulatory filings.
- Scalability and Environmental Compliance: The synthetic route is inherently scalable, moving seamlessly from gram-scale laboratory synthesis to multi-kilogram commercial production. The avoidance of chlorinated solvents in certain steps and the reduction of solvent usage through recrystallization align with green chemistry principles. This environmental compliance is increasingly important for pharmaceutical suppliers who must adhere to strict corporate sustainability goals and regulatory guidelines regarding solvent emissions and hazardous waste management.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the synthesis and application of Carfilzomib Impurity V. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the feasibility and specifications of the method.
Q: Why is synthesizing Carfilzomib Impurity V directly preferred over isolation?
A: Direct synthesis avoids the difficult and low-yield process of isolating trace impurities from the main drug reaction mixture using column chromatography, ensuring higher purity and availability for quality control standards.
Q: What catalysts are used in the preparation of the intermediate Compound II?
A: The process utilizes Palladium on Carbon (Pd/C), available in both wet and dry forms (10% or 20% loading), which allows for efficient hydrogenation under mild temperature conditions.
Q: How is the final purity of Impurity V ensured without chromatography?
A: The method employs a recrystallization step using solvents like dibutyl ketone, acetone, or butanone, which effectively purifies the solid product to high standards suitable for reference materials.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carfilzomib Impurity V Supplier
At NINGBO INNO PHARMCHEM, we understand the critical role that high-quality reference standards play in the drug development lifecycle. Our team of expert chemists is well-versed in the complexities of peptide synthesis and impurity profiling, ensuring that we can deliver Carfilzomib Impurity V with the highest levels of purity and structural confirmation. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, leveraging our state-of-the-art facilities to meet stringent purity specifications. Our rigorous QC labs utilize advanced analytical techniques to verify every batch, guaranteeing that the material you receive is fit for purpose in your regulatory submissions.
We invite you to collaborate with us to secure a stable supply of this essential reference material. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact us today to request specific COA data and route feasibility assessments, and let us support your commitment to delivering safe and effective therapies to patients worldwide.