Advanced Chiral Catalytic Hydrogenation Strategy for Scalable Duvelisib Manufacturing
Advanced Chiral Catalytic Hydrogenation Strategy for Scalable Duvelisib Manufacturing
The pharmaceutical industry is constantly seeking more efficient, cost-effective, and environmentally sustainable pathways for the production of complex oncology therapeutics. Patent CN111635404A introduces a groundbreaking preparation method for Duvelisib, a potent PI3K inhibitor, which fundamentally shifts the synthetic paradigm from traditional chiral pool synthesis to a sophisticated late-stage asymmetric hydrogenation strategy. This novel approach addresses critical bottlenecks in the existing manufacturing landscape by utilizing 2-chloro-6-methyl-N-phenylbenzamide as a robust starting material, thereby bypassing the need for expensive, protected chiral amino acid derivatives that have historically constrained supply chains. By integrating a sequence of oxidation, epoxidation, rearrangement, cyclization, imidization, and finally, chiral catalytic hydrogenation, this technology offers a streamlined route that promises significant improvements in overall yield and operational safety. For global procurement teams and R&D directors, understanding the mechanistic nuances of this pathway is essential for evaluating its potential as a reliable API intermediate supplier solution.
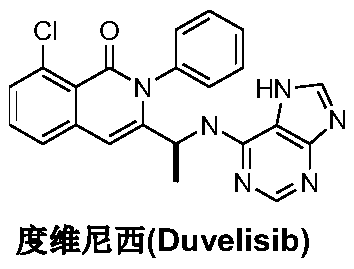
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Duvelisib and its analogs has relied heavily on the construction of a chiral amine parent nucleus, often designated as Intermediate A, which serves as the foundational scaffold for the final molecule. As illustrated in prior art such as international patents WO2011008302A1 and WO2011146882A1, these conventional routes typically commence with 2-chloro-3-methylbenzoic acid or its esters coupled with S-alanine derivatives. While chemically valid, this approach suffers from inherent inefficiencies, including a protracted sequence of condensation, cyclization, protection, and deprotection reactions that drastically increase the step count. The reliance on chiral alanine sources not only inflates the raw material costs due to the premium pricing of enantiopure amino acids but also introduces significant waste streams associated with protecting group manipulation. Furthermore, the harsh reaction conditions often required for these transformations can compromise scalability, leading to lower throughput and increased safety risks in a commercial manufacturing setting.
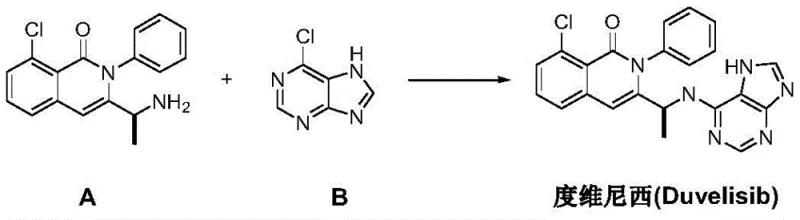
The Novel Approach
In stark contrast to the legacy methodologies, the process disclosed in CN111635404A adopts a convergent strategy that defers the introduction of chirality until the final stages of synthesis. This innovative route begins with the inexpensive and readily available 2-chloro-6-methyl-N-phenylbenzamide, subjecting it to a carefully orchestrated series of transformations including Selectfluor-mediated oxidation and DBU-promoted epoxidation. The true brilliance of this method lies in its ability to construct the complex isoquinolinone core prior to the stereoselective step, thereby minimizing the risk of racemization during earlier high-energy transformations. By employing a specialized iron-catalyzed rearrangement followed by a highly selective asymmetric hydrogenation, this new pathway effectively decouples the structural complexity from the chiral induction step. This strategic decoupling not only simplifies the purification protocols but also enhances the overall atom economy, making it a superior choice for cost reduction in pharmaceutical manufacturing.
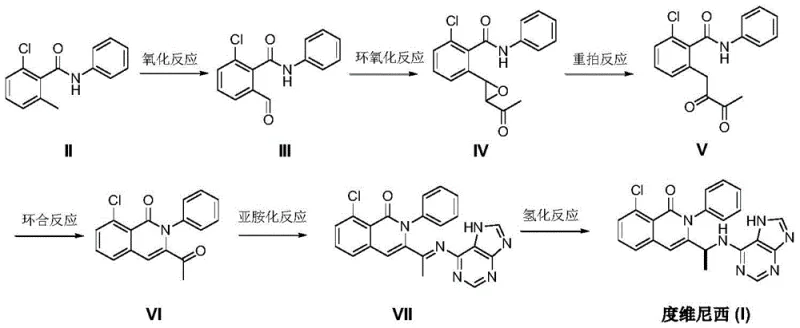
Mechanistic Insights into Iron-Catalyzed Rearrangement and Asymmetric Hydrogenation
The core chemical innovation of this patent resides in the sophisticated application of transition metal catalysis to drive both skeletal rearrangement and stereochemical control. Specifically, the transformation of the epoxy-ketone intermediate (IV) into the 1,2-diketone species (V) is mediated by tetraphenylporphyrin iron triflate [Fe(tpp)OTf]. This Lewis acidic iron catalyst facilitates a precise migration of the acetyl group, a critical step that sets up the subsequent cyclization to form the isoquinolinone ring system. The efficiency of this rearrangement is paramount, as it dictates the formation of the key 3-acetyl-8-chloro-2-phenyl-1(2H)-isoquinolinone intermediate (VI) with high fidelity. Following the imidization with 6-amino-9H-purine to generate the imine intermediate (VII), the process culminates in a highly enantioselective hydrogenation. This final step utilizes a bulky chiral ligand, (S)-6,12-bis(3,5-di-tert-butylphenyl)-9-methylene-9,10-dihydroxy-8H-dinaphtho[2,1-f:1',2'-h][1,5]dioxocin, in conjunction with a bis(pentafluorophenyl)borane cocatalyst. The steric bulk of the tert-butyl groups on the ligand creates a confined chiral environment that directs the hydride delivery exclusively to one face of the imine double bond, ensuring the formation of the desired (S)-enantiomer with exceptional optical purity.
From an impurity control perspective, this mechanistic pathway offers distinct advantages over traditional methods. By avoiding the use of reactive alkylating agents on a chiral center early in the synthesis, the risk of generating diastereomeric impurities is significantly mitigated. The use of mild acidic conditions for the cyclization step (using hydrogen chloride in toluene) and the specific catalytic system for hydrogenation ensures that side reactions such as over-reduction or hydrolysis of the sensitive purine ring are minimized. The patent data indicates that the final hydrogenation step proceeds at moderate temperatures (25-35°C) and pressures (30 bar), conditions that are gentle enough to preserve the integrity of the heterocyclic core while robust enough to drive the reaction to completion. This level of control is crucial for meeting the stringent purity specifications required for oncology drugs, where even trace levels of genotoxic impurities or wrong isomers can lead to batch rejection.
How to Synthesize Duvelisib Efficiently
The synthesis of Duvelisib via this novel route involves a logical progression of six distinct unit operations, each optimized for high conversion and minimal byproduct formation. The process begins with the oxidation of the methyl group on the benzamide ring, followed by epoxidation and a unique iron-catalyzed rearrangement to establish the carbon skeleton necessary for ring closure. Once the isoquinolinone core is formed and coupled with the purine moiety, the final asymmetric hydrogenation installs the critical chiral center. This sequence represents a significant departure from linear syntheses, offering a more convergent and manageable workflow for process chemists. The detailed standardized synthesis steps, including specific reagent ratios, solvent choices, and temperature profiles for each stage, are outlined in the technical guide below to facilitate immediate technology transfer and pilot plant evaluation.
- Oxidation of 2-chloro-6-methyl-N-phenylbenzamide using Selectfluor and ferrous chloride to form the aldehyde intermediate.
- Epoxidation with chloroacetone and DBU followed by iron-catalyzed rearrangement to generate the diketone precursor.
- Cyclization with hydrogen chloride, imidization with 6-amino-9H-purine, and final chiral hydrogenation using a BINOL-derived catalyst.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthesis route translates into tangible strategic benefits that extend beyond simple chemistry. The most immediate impact is observed in the raw material sourcing strategy, as the process eliminates the dependency on volatile and expensive chiral amino acid markets. By switching to commodity chemicals like 2-chloro-6-methyl-N-phenylbenzamide and chloroacetone, manufacturers can secure a more stable and predictable supply base, insulating production schedules from the fluctuations typical of specialty chiral building blocks. Furthermore, the reduction in the total number of synthetic steps directly correlates to a reduction in manufacturing lead time and labor costs. Fewer isolation and purification stages mean less solvent consumption, lower energy usage for heating and cooling, and reduced waste disposal costs, all of which contribute to a leaner and more competitive cost structure for the final API.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the substitution of high-cost chiral starting materials with inexpensive achiral precursors. In traditional routes, the cost of S-alanine derivatives and the reagents required for their protection and deprotection can account for a substantial portion of the COGS. By deferring chirality introduction to the final hydrogenation step using a catalytic amount of chiral ligand, the material cost per kilogram of product is drastically lowered. Additionally, the high yields reported in the patent examples for each step—often exceeding 80% and reaching up to 94% in the oxidation step—minimize material loss and maximize throughput, further enhancing the overall economic efficiency of the manufacturing campaign.
- Enhanced Supply Chain Reliability: Supply chain resilience is significantly bolstered by the use of widely available industrial solvents and reagents throughout the synthesis. Solvents such as toluene, acetonitrile, and dichloromethane are standard commodities with robust global supply networks, reducing the risk of production stoppages due to solvent shortages. Moreover, the reaction conditions are designed to be operationally simple, avoiding the need for cryogenic temperatures below -50°C or ultra-high pressure equipment that might limit the number of qualified contract manufacturing organizations (CMOs). This accessibility allows for greater flexibility in vendor selection and capacity allocation, ensuring consistent delivery of high-purity pharmaceutical intermediates to downstream formulation partners.
- Scalability and Environmental Compliance: From an environmental and safety standpoint, this route aligns well with green chemistry principles, which is increasingly a prerequisite for regulatory approval and corporate sustainability goals. The avoidance of heavy metal catalysts in favor of iron-based systems for the rearrangement step reduces the burden of heavy metal clearance testing and waste treatment. The process operates largely at atmospheric or moderate pressures, and the exothermic nature of the reactions is manageable within standard reactor configurations. This inherent safety profile facilitates easier scale-up from kilogram to multi-ton production scales without requiring extensive re-engineering of the process infrastructure, thereby accelerating the time to market for generic or biosimilar versions of the drug.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Duvelisib synthesis technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing a clear picture of the process capabilities and limitations. Understanding these details is vital for technical teams assessing the feasibility of adopting this route for commercial production.
Q: What is the primary advantage of this new Duvelisib synthesis route?
A: The primary advantage is the elimination of expensive chiral starting materials like S-alanine derivatives. Instead, chirality is introduced late-stage via asymmetric hydrogenation, significantly reducing raw material costs and simplifying the supply chain.
Q: How does the patent ensure high enantiomeric purity?
A: High purity is achieved through the use of a specialized chiral catalyst system in the final hydrogenation step, specifically utilizing an (S)-BINOL-derived ligand配合 with a bis(pentafluorophenyl)borane cocatalyst under controlled pressure and temperature.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process utilizes readily available raw materials and avoids harsh conditions. The reaction temperatures are moderate (mostly below 120°C), and the solvents used are standard industrial grades like toluene and acetonitrile, facilitating easy scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Duvelisib Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this advanced chiral catalytic hydrogenation strategy for the global supply of Duvelisib. As a premier CDMO partner, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent are fully realized in a GMP-compliant manufacturing environment. Our state-of-the-art facilities are equipped to handle the specific requirements of this synthesis, including the safe handling of Selectfluor and the precise control needed for the asymmetric hydrogenation step. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Duvelisib intermediate meets the highest quality standards required by international regulatory bodies.
We invite forward-thinking pharmaceutical companies to collaborate with us to leverage this cost-effective and scalable synthesis route. By partnering with our technical procurement team, you can gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise in complex organic synthesis can optimize your supply chain and accelerate your product development timelines.