Advanced Chemical Synthesis of Nelarabine Intermediates for Commercial Scale-Up
Advanced Chemical Synthesis of Nelarabine Intermediates for Commercial Scale-Up
The pharmaceutical landscape for antineoplastic agents continues to evolve, driven by the need for more efficient and scalable manufacturing processes for critical drugs like nelarabine. As a prodrug of the guanosine analogue 9-β-D-arabinofuranosylguanine (Ara-G), nelarabine plays a pivotal role in the treatment of T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. A recent technological breakthrough detailed in patent CN108373491B introduces a robust chemical preparation method that fundamentally shifts the production paradigm from complex biocatalytic or multi-step configuration conversion routes to a streamlined chemical synthesis. This innovation leverages cyclocytidine hydrochloride and 2-amino-6-chloropurine as primary feedstocks, utilizing a Lewis acid-catalyzed base conversion strategy to achieve high-purity targets. For global supply chain stakeholders, this patent represents a significant opportunity to optimize the commercial scale-up of complex pharmaceutical intermediates, offering a pathway that is not only chemically elegant but also economically superior.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the development of the methodology described in CN108373491B, the industrial synthesis of nelarabine was plagued by significant technical and economic bottlenecks. Historical literature describes several approaches, including bio-enzyme catalysis, which relies on invertase; however, this enzyme is notoriously difficult to prepare, expensive to source, and presents substantial challenges in downstream purification and process amplification. Alternative chemical routes, such as the ribose configuration conversion method disclosed in Chinese patent CN101092441A, involve converting ribofuranose to arabinose through a tedious six-step sequence. These legacy processes suffer from low total yields, harsh reaction conditions, and extreme difficulty in controlling both chemical and optical purity due to the complexity of selective protection and configuration inversion steps. Furthermore, methods starting from vidarabine or D-arabinose often require nitration and reduction steps or multi-step glycosidic bond formations that introduce safety hazards and operational complexities, making them less ideal for large-scale cost reduction in pharmaceutical intermediates manufacturing.
The Novel Approach
The novel approach outlined in the patent circumvents these historical inefficiencies by employing a direct coupling strategy that capitalizes on the inherent stereochemistry of cyclocytidine. By hydrolyzing cyclocytidine hydrochloride to form cytarabine and subsequently protecting it as tetraacetyl cytarabine, the process establishes the correct arabinose configuration early in the synthesis without the need for difficult inversion steps. This intermediate is then coupled with a silanized 2-amino-6-methoxypurine derivative under the catalytic influence of trimethylsilyl trifluoromethanesulfonate (TMSOTf). This route is characterized by its brevity, mild reaction conditions, and the use of inexpensive, readily available raw materials. The elimination of enzymatic steps and the reduction of synthetic linear steps directly translate to a more robust process that is easier to control and safer to operate, addressing the core pain points of yield and purity that have long hindered the efficient production of this critical oncology drug.
Mechanistic Insights into TMSOTf-Catalyzed Glycosylation
The cornerstone of this synthesis is the Lewis acid-catalyzed glycosylation step, which dictates the stereochemical outcome and overall efficiency of the transformation. The mechanism begins with the activation of the purine base; 2-amino-6-methoxypurine is first silylated using trimethylchlorosilane and triethylamine to enhance its nucleophilicity and solubility in organic media. Simultaneously, the sugar donor, tetraacetyl cytarabine, possesses a leaving group at the anomeric position that is activated by the strong Lewis acid TMSOTf. The triflate anion coordinates with the acetoxy group at the C1 position of the sugar, generating a highly reactive oxocarbenium ion intermediate. This electrophilic species is then attacked by the N9 nitrogen of the silylated purine base. The steric environment provided by the C2 acetoxy group on the sugar ring directs the incoming nucleophile to attack from the opposite face, ensuring the exclusive formation of the desired β-anomer. This precise stereocontrol is critical, as the biological activity of nelarabine is strictly dependent on the β-configuration of the glycosidic bond.
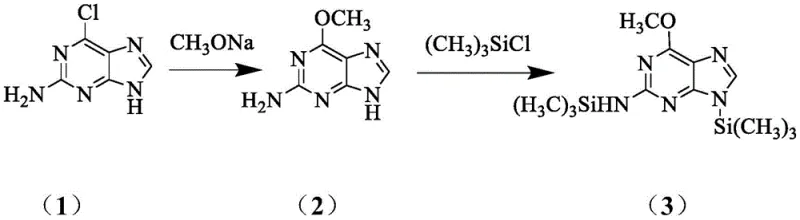
Following the coupling reaction, the resulting protected intermediate undergoes a deprotection sequence to reveal the final active pharmaceutical ingredient. The use of TMSOTf is particularly advantageous because it operates effectively at moderate temperatures (60-84°C), minimizing the risk of thermal degradation of the sensitive purine and sugar moieties. Furthermore, the reaction conditions allow for the use of common organic solvents such as acetonitrile or 1,2-dichloroethane, which facilitates easy workup and purification. The subsequent deacetylation using methanolic ammonia or sodium methoxide cleanly removes the protecting groups without affecting the glycosidic bond integrity. This mechanistic pathway ensures that impurities arising from anomerization or base degradation are kept to a minimum, resulting in a product profile that meets the stringent quality requirements of modern regulatory bodies.
How to Synthesize Nelarabine Efficiently
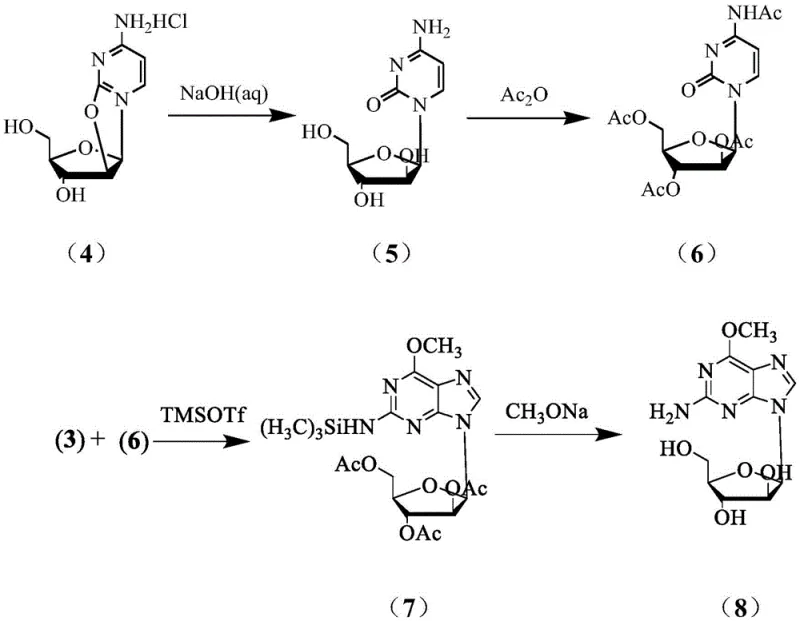
The synthesis of nelarabine via this patented route involves a logical sequence of transformations designed to maximize yield while minimizing operational complexity. The process begins with the methoxylation of 2-amino-6-chloropurine, followed by silylation to prepare the nucleophilic partner. In parallel, cyclocytidine hydrochloride is hydrolyzed to cytarabine and then peracetylated to activate the anomeric center. The convergence of these two streams occurs in the presence of the TMSOTf catalyst, forming the core carbon-nitrogen bond. Finally, global deprotection yields the target molecule. This modular approach allows for the optimization of each step independently, providing process chemists with significant flexibility in troubleshooting and scaling. For detailed operational parameters and stoichiometric ratios, please refer to the standardized synthesis guide below.
- React 2-amino-6-chloropurine with sodium methoxide in methanol at reflux (67°C) to form 2-amino-6-methoxypurine.
- Silylate the methoxypurine using trimethylchlorosilane and triethylamine in an organic solvent like 1,2-dichloroethane.
- Hydrolyze cyclocytidine hydrochloride in aqueous alkali (pH 12-14) to obtain cytarabine, followed by acetylation with acetic anhydride.
- Couple the silylated purine and acetylated cytarabine using TMSOTf catalyst at 60-84°C to form the protected nelarabine intermediate.
- Deprotect the intermediate using methanolic ammonia or sodium methoxide to yield the final nelarabine product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of the synthesis method described in CN108373491B offers compelling strategic advantages that extend beyond mere technical feasibility. The shift from enzyme-dependent or multi-step configuration inversion routes to this direct chemical coupling fundamentally alters the cost structure and risk profile of nelarabine production. By relying on stable, commodity-grade chemicals rather than specialized biocatalysts, manufacturers can secure a more resilient supply chain that is less susceptible to biological variability or sourcing bottlenecks. The simplified workflow reduces the number of unit operations, which in turn lowers capital expenditure requirements for reactor volume and purification infrastructure. This efficiency gain is critical for maintaining competitiveness in the generic oncology market, where margin pressure is intense and speed to market is paramount.
- Cost Reduction in Manufacturing: The most immediate impact of this technology is the drastic reduction in raw material costs. Cyclocytidine hydrochloride and 2-amino-6-chloropurine are significantly cheaper and more accessible than the specialized enzymes or protected ribose derivatives required in alternative methods. Furthermore, the elimination of the invertase enzyme removes the need for costly fermentation, extraction, and stabilization processes associated with biocatalysts. The reaction conditions are mild enough to reduce energy consumption, and the high selectivity of the TMSOTf-catalyzed step minimizes the loss of valuable intermediates to side reactions. Qualitatively, this translates to a substantially lower cost of goods sold (COGS), enabling manufacturers to offer more competitive pricing or enjoy healthier margins without compromising on quality standards.
- Enhanced Supply Chain Reliability: Supply chain continuity is often jeopardized by reliance on single-source biological reagents or complex chiral starting materials. This chemical route mitigates those risks by utilizing a diverse portfolio of standard chemical reagents that are widely available from multiple global suppliers. The robustness of the chemical steps means that production schedules are more predictable and less prone to the batch-to-batch variability often seen in enzymatic processes. Additionally, the shorter synthetic timeline reduces the working capital tied up in inventory, allowing for a more agile response to market demand fluctuations. This reliability is essential for securing long-term contracts with major pharmaceutical companies that prioritize consistent delivery performance.
- Scalability and Environmental Compliance: From an environmental and safety perspective, this process is inherently greener and safer to scale. The avoidance of harsh nitration reagents and the use of moderate temperatures reduce the potential for runaway reactions and hazardous waste generation. The solvents employed, such as ethyl acetate and acetonitrile, are well-understood and easily recovered, facilitating compliance with increasingly strict environmental regulations regarding volatile organic compound (VOC) emissions. The simplicity of the workup procedures, involving standard filtration and extraction techniques, means that the process can be scaled from pilot plant to multi-ton commercial production with minimal engineering hurdles. This scalability ensures that the technology remains viable as production volumes increase to meet global therapeutic demand.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this nelarabine synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a clear understanding of the process capabilities. Understanding these nuances is vital for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the value proposition of this manufacturing route.
Q: Why is the cyclocytidine route preferred over enzymatic methods for nelarabine production?
A: The cyclocytidine route avoids the use of expensive and difficult-to-prepare invertase enzymes required in bio-catalytic methods. It utilizes cheap, commercially available raw materials and offers a shorter synthetic route with milder reaction conditions, significantly simplifying purification and scale-up.
Q: What is the specific role of TMSOTf in the glycosylation step?
A: Trimethylsilyl trifluoromethanesulfonate (TMSOTf) acts as a potent Lewis acid catalyst. It activates the anomeric center of the tetraacetyl cytarabine, facilitating a highly stereoselective nucleophilic attack by the silylated purine base, which ensures the correct beta-configuration essential for biological activity.
Q: How does this process address impurity control compared to ribose configuration conversion methods?
A: Unlike ribose configuration conversion methods which involve complex selective protection and configuration inversion steps prone to stereochemical errors, this direct coupling method starts with the correct arabinose configuration inherent in cyclocytidine. This drastically reduces the formation of diastereomeric impurities and simplifies the final purification process.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Nelarabine Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and proven manufacturing capabilities. Our team has extensively analyzed the pathway described in CN108373491B and possesses the requisite knowledge to execute this TMSOTf-catalyzed synthesis with precision. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent are fully realized in a GMP-compliant environment. Our rigorous QC labs and stringent purity specifications guarantee that every batch of nelarabine intermediate meets the highest international standards, providing our partners with the confidence needed to advance their clinical and commercial programs.
We invite forward-thinking pharmaceutical companies to collaborate with us to leverage this advanced synthesis route. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and supply chain constraints. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you secure a reliable supply of high-quality nelarabine intermediates, driving down costs and accelerating your time to market in the competitive oncology sector.