Scalable Synthesis of Halogenated Benzothiazepine Intermediates for Advanced Antiviral Drug Development
Introduction to Novel Halogenated Benzothiazepine Synthesis
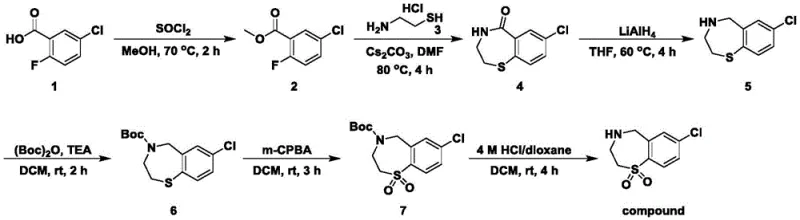
The pharmaceutical industry is constantly seeking robust and scalable pathways for synthesizing complex heterocyclic intermediates, particularly those destined for antiviral applications. A significant breakthrough in this domain is documented in patent CN111548325A, which outlines a sophisticated preparation method for halogenated benzothiazepine compounds. This technology specifically targets the synthesis of 7-chloro-2,3,4,5-tetrahydro-1,4-benzothiazepine-1,1-dioxide, a critical scaffold for developing active components against Respiratory Syncytial Virus (RSV). Unlike traditional methods that often struggle with late-stage functionalization, this approach leverages 5-chloro-2-fluorobenzoic acid as a strategic starting material. By integrating the halogen moiety at the very beginning of the synthetic sequence, the process ensures superior control over the substitution pattern and minimizes the risk of regio-isomer formation. This strategic design not only enhances the chemical integrity of the final product but also streamlines the purification protocols required for GMP manufacturing. For R&D directors and procurement specialists, understanding this pathway offers a glimpse into a more efficient supply chain for high-value antiviral intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of benzothiazepine derivatives has relied heavily on routes that introduce the sulfur and nitrogen atoms separately or utilize less stable precursors. For instance, prior art such as CN106414436A describes a method starting with sodium thiophenolate reacting with 2-chloroethylamine hydrochloride. While feasible, this conventional approach often necessitates multiple protection and deprotection steps to manage the reactivity of the amine and thiol groups simultaneously. Furthermore, introducing halogen substituents onto the benzene ring in these traditional sequences typically requires harsh electrophilic aromatic substitution conditions or expensive cross-coupling catalysts at a later stage. These late-stage modifications frequently lead to lower overall yields due to the accumulation of impurities and the degradation of sensitive heterocyclic cores. The reliance on strong bases and high temperatures in older methodologies can also compromise the stereochemical integrity of the molecule, creating significant challenges for downstream processing and regulatory approval in pharmaceutical manufacturing.
The Novel Approach
In stark contrast, the methodology disclosed in the present patent revolutionizes the construction of the benzothiazepine core by employing a convergent strategy centered on nucleophilic aromatic substitution. By utilizing 5-chloro-2-fluorobenzoic acid, the process capitalizes on the activating effect of the ester group to facilitate the displacement of the fluorine atom by the thiol nucleophile. This intramolecular cyclization is highly efficient and occurs under relatively mild conditions compared to classical Friedel-Crafts type reactions. The sequential order of operations—esterification, cyclization, reduction, protection, oxidation, and deprotection—is meticulously optimized to preserve the halogen functionality throughout the synthesis. This approach eliminates the need for difficult post-synthetic halogenation, thereby reducing the number of unit operations and the associated solvent waste. For supply chain managers, this translates to a more predictable production timeline and a reduced dependency on scarce catalysts, ensuring a steady flow of high-purity intermediates for drug development programs.
Mechanistic Insights into Nucleophilic Substitution and Selective Oxidation
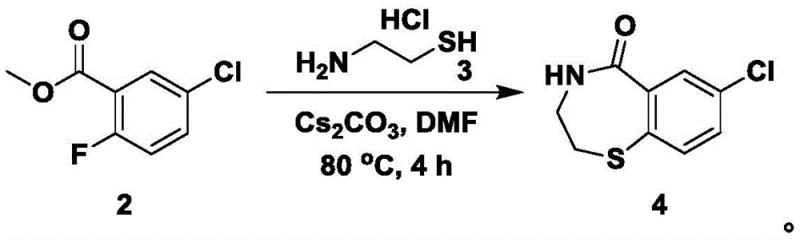
The cornerstone of this synthetic route lies in the mechanistic elegance of the ring-closing step (Step S2), where the interplay between the nucleophile and the electrophilic aromatic system is finely tuned. In this stage, the methyl ester of 5-chloro-2-fluorobenzoic acid reacts with aminoethanethiol hydrochloride. The presence of cesium carbonate is critical here; it serves a dual purpose as both a base to deprotonate the thiol and a scavenger for the hydrofluoric acid byproduct. The generation of carbon dioxide gas during this neutralization helps drive the equilibrium forward, ensuring high conversion rates without the need for excessive reagent loading. Following the formation of the thioether linkage, the pendant amine attacks the ester carbonyl to close the seven-membered ring, forming the 7-chloro-3,4-dihydrobenzo-1,4-thiazepin-5(2H)-one scaffold. This cascade reaction is a prime example of atom economy, constructing two bonds in a single operational sequence while maintaining the integrity of the chloro-substituent.
Furthermore, the selective oxidation of the sulfur atom in Step S5 demonstrates precise control over chemoselectivity. Using meta-chloroperoxybenzoic acid (m-CPBA) as the oxidant allows for the transformation of the sulfide into the corresponding sulfone (1,1-dioxide) without affecting the protected amine or the aromatic chloride. This selectivity is paramount because over-oxidation or N-oxidation would generate difficult-to-remove impurities that could compromise the safety profile of the final drug substance. The reaction proceeds smoothly at room temperature, indicating a low activation energy barrier and high reagent efficiency. For quality control teams, this mild condition minimizes the formation of thermal degradation products, resulting in a cleaner crude profile that simplifies the final crystallization or chromatographic purification steps. The mechanistic robustness of these key transformations underpins the reliability of the entire manufacturing process.
How to Synthesize 7-chloro-2,3,4,5-tetrahydro-1,4-benzothiazepine-1,1-dioxide Efficiently
Executing this synthesis requires strict adherence to the optimized parameters regarding temperature, stoichiometry, and reaction time to maximize yield and purity. The process begins with the activation of the carboxylic acid followed by a telescoped cyclization that sets the core structure. Subsequent reduction of the ketone and protection of the secondary amine prepares the molecule for the critical oxidation step. Each stage is monitored via TLC or LCMS to ensure complete conversion before proceeding, preventing the carryover of reactive intermediates that could complicate downstream workups. The final deprotection utilizes acidic conditions compatible with the sulfone moiety, yielding the target free amine as a stable hydrochloride salt or free base depending on the workup. For detailed operational procedures, including specific solvent volumes and quenching protocols, please refer to the standardized guide below.
- Esterify 5-chloro-2-fluorobenzoic acid with methanol using thionyl chloride at 70°C to form the methyl ester.
- Perform nucleophilic substitution and ring closure with aminoethanethiol hydrochloride using cesium carbonate in DMF at 80°C.
- Reduce the ketone carbonyl using lithium aluminum hydride in THF, followed by Boc protection of the amine.
- Oxidize the sulfur atom to a sulfone using m-CPBA in dichloromethane at room temperature.
- Remove the Boc protecting group using 4M HCl in dioxane to yield the final halogenated benzothiazepine compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented synthesis route offers substantial advantages that directly impact the bottom line and supply chain resilience for pharmaceutical manufacturers. The shift away from transition metal catalysts and harsh halogenation reagents significantly reduces the cost of goods sold (COGS) by eliminating expensive catalyst recovery processes and specialized waste treatment requirements. The use of commodity chemicals like thionyl chloride, cesium carbonate, and m-CPBA ensures that raw material sourcing remains stable and unaffected by geopolitical fluctuations often seen with precious metals. Moreover, the high atom economy of the cyclization step means less raw material is wasted as byproduct, further enhancing the economic viability of large-scale production. For procurement managers, this translates into a more predictable pricing model and reduced risk of supply disruption for critical intermediates.
- Cost Reduction in Manufacturing: The elimination of late-stage halogenation steps removes the need for costly palladium or copper catalysts and the associated ligand systems. This simplification of the synthetic tree reduces the number of purification columns required, lowering solvent consumption and energy usage per kilogram of product. Additionally, the high yields reported in the patent examples suggest that less starting material is needed to produce the same amount of API intermediate, driving down the overall material cost significantly.
- Enhanced Supply Chain Reliability: By relying on robust, non-proprietary reagents such as 5-chloro-2-fluorobenzoic acid, manufacturers can diversify their supplier base and avoid single-source bottlenecks. The process operates under standard pressure and moderate temperatures, meaning it can be executed in existing multipurpose reactors without requiring specialized high-pressure or cryogenic equipment. This flexibility allows for rapid scale-up from pilot plant to commercial tonnage, ensuring that clinical trial demands can be met without lengthy lead times.
- Scalability and Environmental Compliance: The workflow generates manageable byproducts like carbon dioxide and salts that are easier to treat than heavy metal waste streams. The use of dichloromethane and DMF, while requiring careful handling, is well-established in industrial settings with mature recovery protocols. The overall reduction in step count compared to legacy routes decreases the total volume of hazardous waste generated, aligning with modern green chemistry principles and facilitating smoother regulatory audits for environmental compliance.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis pathway. These insights are derived directly from the experimental data and mechanistic rationale provided in the patent documentation. Understanding these nuances is essential for process chemists aiming to transfer this technology from the laboratory to the production floor. The answers highlight the critical control points that ensure consistent product quality and batch-to-batch reproducibility.
Q: What is the primary advantage of using 5-chloro-2-fluorobenzoic acid as the starting material?
A: Using 5-chloro-2-fluorobenzoic acid allows for the early introduction of the halogen substituent on the benzene ring, which simplifies the synthetic route compared to post-synthesis halogenation and improves the overall stability and yield of the intermediate.
Q: Why is cesium carbonate preferred over other bases in the cyclization step?
A: Cesium carbonate acts as an effective base that scavenges the hydrofluoric acid byproduct generated during the nucleophilic substitution, driving the reaction equilibrium forward and preventing side reactions associated with acidic conditions.
Q: How does this method ensure high purity for pharmaceutical applications?
A: The process utilizes mild reaction conditions and specific reagents like m-CPBA for selective oxidation, minimizing impurity formation. Additionally, the final step employs medium-pressure liquid chromatography to ensure stringent purity specifications required for active pharmaceutical ingredients.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 7-chloro-2,3,4,5-tetrahydro-1,4-benzothiazepine-1,1-dioxide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of antiviral therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and speed. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of 7-chloro-2,3,4,5-tetrahydro-1,4-benzothiazepine-1,1-dioxide meets the highest industry standards. Our commitment to technical excellence allows us to navigate complex synthetic challenges efficiently, providing our partners with a secure and reliable supply chain foundation.
We invite you to collaborate with us to leverage this advanced synthesis technology for your RSV drug development programs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your timeline to market while optimizing your overall production costs.