Advanced Synthesis of 8-Chloro-Benzothiazepine Oxide for Scalable Pharmaceutical Manufacturing
Introduction to Next-Generation Benzothiazepine Oxide Synthesis
The pharmaceutical industry is constantly seeking robust synthetic routes for heterocyclic intermediates that serve as the backbone for potent antiviral therapies. A significant breakthrough in this domain is detailed in patent CN111518055A, which discloses a novel preparation method for benzothiazepine oxide, specifically targeting the synthesis of 8-chloro-2,3,4,5-tetrahydro-1,4-benzothiazepine-1,1-dioxide. This compound acts as a critical intermediate for active pharmaceutical ingredients (APIs) designed to prevent and treat Respiratory Syncytial Virus (RSV) infections. The technical innovation lies not merely in the creation of the heterocycle but in the strategic and efficient introduction of a chlorine atom at the 8-position of the benzene ring, a modification known to significantly enhance pharmacological properties such as metabolic stability and binding affinity.
For R&D directors and process chemists, the value of this patent extends beyond the final molecule; it offers a comprehensive, nine-step synthetic pathway that balances chemical complexity with operational feasibility. By utilizing 2-fluoro-4-chlorobenzoic acid as the starting material, the method circumvents the difficulties associated with late-stage halogenation, which often plagues traditional heterocyclic synthesis. This approach ensures that the chlorine substituent is embedded early in the molecular architecture, providing a stable foundation for subsequent transformations including esterification, thioether formation, cyclization, and oxidation. The result is a highly defined impurity profile and a scalable process suitable for commercial manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of benzothiazepine derivatives has relied on pathways that struggle with regioselective halogenation. Prior art, such as the methods disclosed in CN109879837A, often employs thiophenol sodium reacting with chloroethylamines to build the side chain before attempting to construct the ring system. A major drawback of these conventional routes is the difficulty in introducing specific substituents, like chlorine, onto the aromatic ring without affecting other sensitive functional groups or requiring harsh conditions that degrade yield. Furthermore, traditional methods may involve multiple protection and deprotection cycles that add unnecessary steps, increase waste generation, and complicate the purification process, ultimately driving up the cost of goods sold (COGS) and extending the lead time for high-purity pharmaceutical intermediates.
The Novel Approach
The methodology presented in CN111518055A revolutionizes this landscape by reversing the synthetic logic. Instead of struggling to chlorinate a pre-formed ring, the process begins with a chlorinated benzoic acid derivative. This strategic shift allows for the direct construction of the chlorinated scaffold. The route proceeds through a logical sequence: protection of the amine, esterification of the acid, nucleophilic aromatic substitution to install the sulfur linker, and finally, ring closure and oxidation. This linear progression minimizes cross-reactivity and maximizes atom economy at each stage.
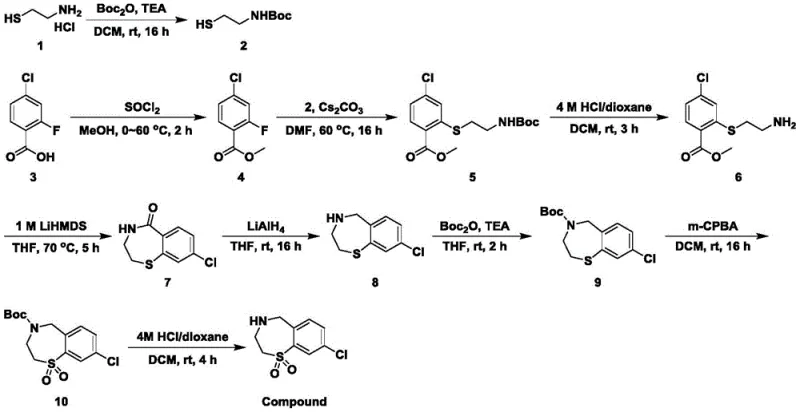
As illustrated in the reaction scheme above, the pathway integrates modern organic synthesis techniques, such as the use of cesium carbonate for efficient fluoride displacement and lithium aluminum hydride for selective carbonyl reduction. For procurement managers, this translates to a reliable pharmaceutical intermediate supplier capability, as the starting materials are commercially available commodity chemicals, reducing supply chain volatility. The clarity of this route facilitates easier technology transfer and scale-up, addressing a critical pain point in the commercialization of complex antiviral drugs.
Mechanistic Insights into Fluoride Displacement and Cyclization
From a mechanistic perspective, the success of this synthesis hinges on two pivotal transformations: the nucleophilic aromatic substitution (Step S3) and the intramolecular cyclization (Step S5). In Step S3, the reaction between methyl 4-chloro-2-fluorobenzoate and the protected aminoethanethiol is driven by the high electronegativity of the fluorine atom ortho to the ester group. The use of cesium carbonate (Cs2CO3) as the base is particularly ingenious; it not only deprotonates the thiol to generate the reactive thiolate nucleophile but also scavenges the hydrogen fluoride byproduct by forming insoluble cesium fluoride. This removal of HF pushes the chemical equilibrium forward, significantly improving conversion rates and minimizing the formation of hydrolysis byproducts that could compromise the purity of the intermediate.
The subsequent ring-closure step (Step S5) employs lithium bis(trimethylsilyl)amide (LiHMDS), a non-nucleophilic strong base, to effect an intramolecular condensation between the primary amine and the methyl ester. This transformation constructs the seven-membered 1,4-benzothiazepine ring. The choice of LiHMDS is critical because it is strong enough to deprotonate the amine without attacking the ester carbonyl directly, thereby favoring the desired cyclization over intermolecular oligomerization. Following this, the oxidation of the sulfur atom in Step S8 using m-chloroperoxybenzoic acid (m-CPBA) converts the thioether into a sulfone. This oxidation state change is essential for the biological activity of the final RSV inhibitor, as the sulfone moiety acts as a strong electron-withdrawing group that modulates the electronic properties of the heterocycle.
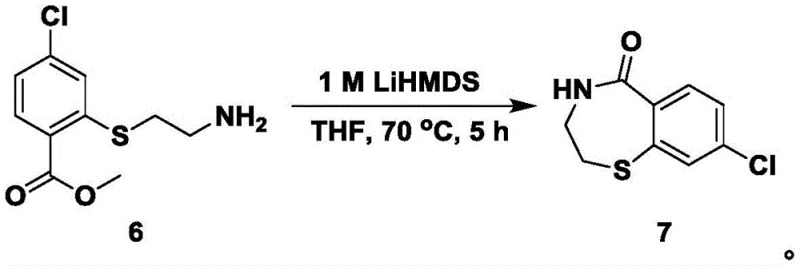
Understanding these mechanistic nuances is vital for quality control teams aiming to maintain high-purity benzothiazepine oxide specifications. By controlling the stoichiometry of the base and the temperature profile during the cyclization, manufacturers can suppress side reactions such as epimerization or over-alkylation. This level of process control ensures that the final API meets the stringent regulatory requirements for antiviral medications, reinforcing the value proposition for partners seeking cost reduction in API manufacturing through improved first-pass yields and reduced rework.
How to Synthesize 8-Chloro-Benzothiazepine Oxide Efficiently
Implementing this synthesis requires precise adherence to the reaction conditions outlined in the patent to ensure reproducibility and safety. The process is divided into nine distinct operational units, ranging from simple acid-base reactions to more complex reductions and oxidations. Key to the success of the operation is the management of exothermic events, particularly during the addition of thionyl chloride in the esterification step and the quenching of the LiHMDS reaction. Detailed standard operating procedures (SOPs) should be established for each unit operation, with particular attention paid to the purification methods, which primarily utilize liquid-liquid extraction and silica gel chromatography to isolate intermediates.
- Protect aminoethanethiol hydrochloride with Boc2O to form tert-butyloxycarbonylaminoethanethiol.
- Esterify 2-fluoro-4-chlorobenzoic acid with methanol and thionyl chloride to form the methyl ester.
- Perform nucleophilic substitution with cesium carbonate to introduce the thioether linkage.
- Deprotect the amine group using HCl/dioxane to reveal the primary amine.
- Cyclize using LiHMDS to form the benzothiazepine ketone ring structure.
- Reduce the ketone carbonyl to a methylene group using lithium aluminum hydride.
- Re-protect the secondary amine with Boc2O prior to oxidation.
- Oxidize the sulfur atom to a sulfone using m-CPBA.
- Final deprotection with HCl/dioxane yields the target benzothiazepine oxide.
Commercial Advantages for Procurement and Supply Chain Teams
For supply chain leaders and procurement specialists, the adoption of this synthetic route offers tangible strategic benefits beyond mere chemical elegance. The primary advantage lies in the robustness of the supply chain for raw materials. By utilizing 2-fluoro-4-chlorobenzoic acid as the starting block, the process relies on widely available bulk chemicals rather than exotic, custom-synthesized precursors. This reduces the risk of supply disruption and provides leverage in price negotiations with upstream vendors. Furthermore, the modular nature of the nine-step synthesis allows for flexible manufacturing strategies, where certain intermediates can be stockpiled or outsourced to specialized contract manufacturers, thereby optimizing inventory levels and working capital.
- Cost Reduction in Manufacturing: The process achieves cost efficiency not through a single magic bullet but through the cumulative effect of optimized yields at each step. The use of cesium carbonate, while initially appearing more expensive than potassium carbonate, actually drives higher conversion in the fluorine displacement step, reducing the amount of unreacted starting material that needs to be recovered or discarded. Additionally, the ability to perform several steps, such as the amine protection and oxidation, under mild room temperature conditions significantly lowers energy consumption compared to processes requiring cryogenic cooling or high-pressure heating, leading to substantial operational expenditure savings.
- Enhanced Supply Chain Reliability: The synthetic route is designed with scalability in mind. The reagents used, such as thionyl chloride, lithium aluminum hydride, and m-CPBA, are standard industrial chemicals with well-established global supply networks. This eliminates the dependency on single-source suppliers for niche catalysts. Moreover, the purification techniques described, primarily extraction and crystallization, are easily transferable from laboratory glassware to large-scale stainless steel reactors, ensuring that the transition from pilot plant to commercial production is smooth and predictable, effectively reducing lead time for high-purity intermediates.
- Scalability and Environmental Compliance: Environmental, Health, and Safety (EHS) considerations are increasingly critical in pharmaceutical manufacturing. This process demonstrates a commitment to greener chemistry by minimizing the use of heavy metal catalysts, which often require complex and costly removal steps to meet residual metal specifications in the final drug product. The waste streams generated are primarily organic solvents and inorganic salts, which can be managed through standard distillation and wastewater treatment protocols. This simplifies the environmental compliance burden and supports the company's sustainability goals, making the commercial scale-up of complex heterocycles more viable in regulated jurisdictions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of 8-chloro-benzothiazepine oxide. These insights are derived directly from the experimental data and technical specifications provided in the patent documentation, offering clarity on process capabilities and product quality attributes.
Q: What is the key advantage of this synthesis route for chlorinated benzothiazepines?
A: The primary advantage is the strategic introduction of the chlorine element at the 8-position early in the synthesis using 2-fluoro-4-chlorobenzoic acid. This avoids complex late-stage halogenation steps that often suffer from poor regioselectivity, ensuring a cleaner impurity profile and higher overall process reliability for antiviral drug development.
Q: How does the process manage safety and scalability concerns with strong bases?
A: The process utilizes lithium bis(trimethylsilyl)amide (LiHMDS) for the critical ring-closure step. While LiHMDS is a strong base, the protocol specifies controlled addition and temperature management (heating to 70°C followed by quenching at 0°C), which mitigates exothermic risks and allows for safe scaling in standard stainless steel reactors commonly found in CDMO facilities.
Q: Why is the sulfur oxidation step performed after amine protection?
A: Oxidizing the sulfur to a sulfone (using m-CPBA) after protecting the amine with a Boc group prevents potential side reactions where the oxidant might attack the free amine nitrogen. This sequence ensures chemoselectivity, preserving the integrity of the nitrogen center while efficiently converting the thioether to the desired sulfone functionality required for biological activity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 8-Chloro-Benzothiazepine Oxide Supplier
At NINGBO INNO PHARMCHEM, we understand that the development of antiviral therapeutics requires a partner who can navigate the complexities of heterocyclic chemistry with precision and reliability. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from clinical trials to market launch. We are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of 8-chloro-benzothiazepine oxide meets the exacting standards required for downstream API synthesis. Our commitment to quality is backed by a robust analytical framework that monitors critical quality attributes at every stage of the manufacturing process.
We invite you to collaborate with us to optimize your supply chain for RSV drug development. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. By leveraging our expertise in process optimization and our established infrastructure, we can help you secure a stable supply of this critical intermediate. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our technical prowess can accelerate your path to commercial success.