Advanced Enantioselective Synthesis of HMG-CoA Reductase Inhibitors for Commercial Scale Manufacturing
Introduction to Advanced Statin Intermediate Manufacturing
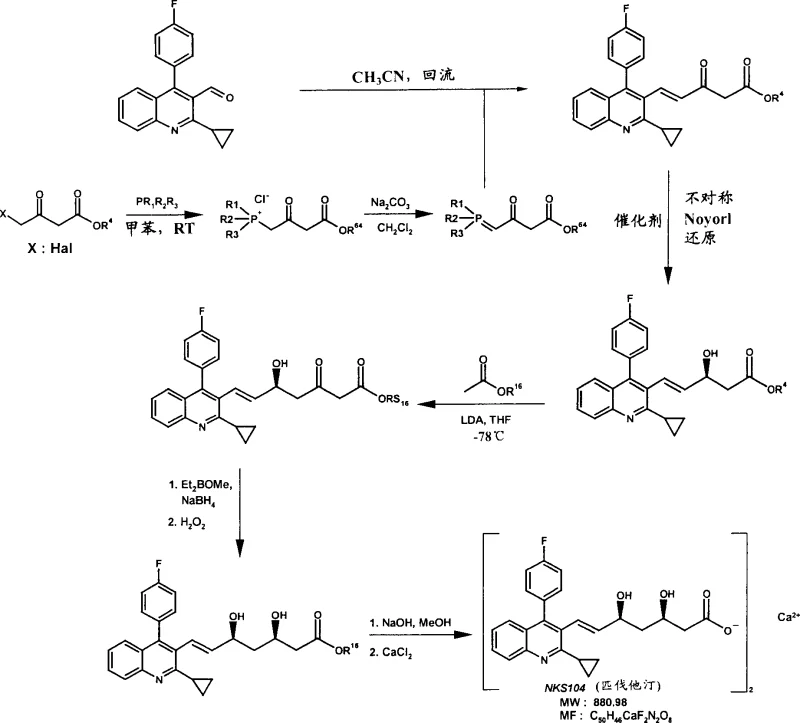
The global demand for HMG-CoA reductase inhibitors, commonly known as statins, continues to drive innovation in pharmaceutical intermediate manufacturing. Patent CN1636004A presents a groundbreaking methodology for the production of mevalonic acid derivatives, which serve as the critical side-chain precursors for major drugs such as Atorvastatin, Fluvastatin, and Pitavastatin. This technology addresses the longstanding challenges associated with establishing the crucial 3,5-cis-diol stereochemistry found in these active pharmaceutical ingredients. By leveraging asymmetric transfer hydrogenation, the disclosed process eliminates the inefficiencies of traditional racemic synthesis followed by resolution. For R&D directors and procurement specialists, understanding this shift from stoichiometric chiral auxiliaries to catalytic enantioselective processes is vital for securing a competitive supply chain. The ability to produce high-purity intermediates with ≥98% enantiomeric excess (ee) directly impacts the regulatory filing success and cost structure of the final drug product.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of the mevalonate side chain has relied heavily on methods that generate racemic mixtures, necessitating a subsequent resolution step to isolate the biologically active enantiomer. As noted in the background of the patent, prior art methods for producing Pitavastatin often involved forming a racemic erythro-β,δ-dihydroxy ester. To obtain the desired optical purity, manufacturers were forced to use chiral resolving agents, such as α-methylbenzylamine, to form diastereomeric salts. The fundamental economic and environmental flaw in this approach is the theoretical maximum yield of 50%, as the unwanted enantiomer is typically discarded or requires complex recycling procedures. Furthermore, these resolution processes often involve multiple crystallization steps, extensive solvent usage, and significant energy consumption for heating and cooling cycles. For a supply chain manager, this translates to higher raw material costs, increased waste disposal liabilities, and longer lead times due to the additional unit operations required to achieve the necessary purity specifications.
The Novel Approach
The process described in CN1636004A revolutionizes this landscape by introducing a direct enantioselective synthetic route. Instead of creating a mixture and separating it, the method constructs the chiral centers directly with high fidelity. The core innovation lies in the use of asymmetric transfer hydrogenation (ATH) utilizing chiral Ruthenium(II) catalysts. This allows for the conversion of prochiral ketones directly into the desired chiral alcohols with exceptional stereocontrol. The patent highlights that this method can achieve an enantiomeric excess of ≥95%, preferably ≥98%, and most preferably ≥99%. By bypassing the resolution step entirely, the process inherently doubles the potential yield from the starting materials compared to resolution-based routes. Additionally, the reaction conditions are remarkably mild, often proceeding at room temperature or moderate heating, which reduces the thermal load on manufacturing equipment. This streamlined approach not only enhances cost reduction in pharmaceutical intermediate manufacturing but also aligns with green chemistry principles by minimizing waste generation and solvent consumption.
Mechanistic Insights into Asymmetric Transfer Hydrogenation
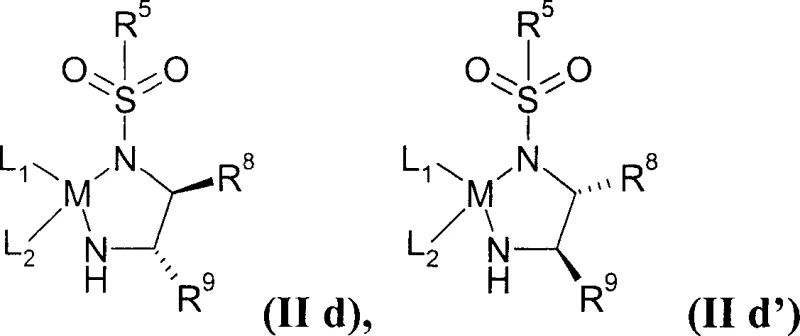
The heart of this technological advancement is the catalytic cycle involving Ru(II) complexes, often referred to as Noyori-type catalysts. The patent specifies the use of catalysts having the general formula (IId), where the metal center is coordinated with chiral diamine ligands and arene groups. Mechanistically, these catalysts operate via a metal-ligand bifunctional pathway. The hydride is transferred from the metal center to the carbonyl carbon of the substrate, while simultaneously, a proton is transferred from the nitrogen of the ligand to the carbonyl oxygen. This concerted mechanism ensures high stereoselectivity without the need for high-pressure hydrogen gas, which poses significant safety and infrastructure challenges in large-scale plants. The patent surprisingly notes that this transfer hydrogenation can be conducted in aqueous solvent systems and even in the absence of inert gas protection, which is a significant departure from the strict anhydrous conditions typically required for organometallic catalysis. This robustness simplifies the operational requirements for commercial scale-up.
Impurity control is another critical aspect addressed by this mechanism. In traditional routes, the formation of syn-diols versus anti-diols is a common selectivity issue. The specific geometry of the Ru-catalyst complex dictates the facial selectivity of the hydride attack, ensuring the formation of the 3,5-cis-diol configuration essential for biological activity. The patent details that the quinoline moiety present in Pitavastatin precursors, which is known to potentially poison hydrogenation catalysts by coordinating to the metal center, does not deactivate the specific Ru(II) catalysts employed in this invention. This tolerance allows for the direct functionalization of complex heterocyclic cores without the need for protecting groups on the nitrogen atoms, further shortening the synthetic sequence. The ability to maintain catalyst activity in the presence of potentially coordinating functional groups ensures consistent reaction rates and prevents the accumulation of unreacted starting materials that could complicate downstream purification.
How to Synthesize Pitavastatin Intermediates Efficiently
The synthesis protocol outlined in the patent provides a clear roadmap for producing the key side-chain intermediates. The process begins with a Wittig olefination to establish the carbon backbone, followed by the critical asymmetric reduction step. Subsequent steps involve aldol condensation to extend the chain and a second stereoselective reduction to install the distal hydroxyl group. The detailed standard operating procedures for each reaction stage, including specific solvent choices like tetrahydrofuran and acetonitrile, and temperature controls ranging from -78°C to reflux, are essential for reproducibility. For technical teams looking to implement this technology, adherence to the specified molar ratios of hydrogen donors, such as formic acid and triethylamine, is crucial for maximizing turnover numbers. The following guide summarizes the critical operational parameters derived from the working examples to ensure successful technology transfer.
- Perform Wittig reaction between a phosphonium ylide and a cyclic aldehyde to form an alpha,beta-unsaturated ketone intermediate.
- Execute asymmetric transfer hydrogenation using a chiral Ru(II) catalyst and a hydrogen donor like formic acid/triethylamine or 2-propanol to establish the first chiral center.
- Conduct aldol condensation with a protected acetate equivalent followed by stereoselective reduction of the resulting beta-keto ester to install the second hydroxyl group.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this enantioselective process offers tangible strategic benefits beyond simple yield improvements. The elimination of the chiral resolution step removes a major bottleneck in production capacity. Traditional resolution requires large volumes of solvents for crystallization and significant reactor occupancy time; removing this step frees up assets and reduces the overall cycle time per batch. Furthermore, the catalysts described, while sophisticated, are used in relatively low loadings and can often be recovered or scavenged, mitigating the cost impact of precious metals. The robustness of the reaction conditions, specifically the tolerance to moisture and air, lowers the barrier for entry for contract manufacturing organizations (CMOs) that may not have specialized high-pressure hydrogenation suites. This flexibility expands the pool of potential suppliers, enhancing supply chain reliability and reducing the risk of single-source dependency.
- Cost Reduction in Manufacturing: The primary driver for cost optimization in this process is the dramatic improvement in atom economy. By avoiding the destruction of 50% of the material inherent in resolution processes, the effective cost of goods sold (COGS) for the active intermediate is significantly lowered. Additionally, the reduction in unit operations—specifically the removal of salt formation, filtration, and regeneration steps—leads to substantial savings in labor, utilities, and waste treatment. The use of transfer hydrogenation avoids the capital expenditure associated with high-pressure hydrogen reactors, allowing for production in standard glass-lined or stainless steel vessels. These factors combine to create a more economically attractive manufacturing profile that can withstand market price pressures.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by the complexity of multi-step syntheses. This streamlined route reduces the number of intermediate isolation points, thereby decreasing the probability of yield loss or quality failure at any single stage. The starting materials, such as the cyclic aldehydes and phosphonium salts, are commercially available or easily synthesized from commodity chemicals, ensuring a stable upstream supply. Moreover, the ability to run reactions under less stringent atmospheric conditions reduces the risk of batch failures due to minor leaks or moisture ingress, leading to more predictable delivery schedules. This reliability is paramount for maintaining the inventory levels required by just-in-time pharmaceutical manufacturing models.
- Scalability and Environmental Compliance: Scaling chemical processes often reveals hidden hazards or inefficiencies, but the chemistry described here is inherently scalable. The exothermic nature of the reactions is manageable, and the solvents used are standard industrial grades. From an environmental perspective, the process generates significantly less waste, particularly organic salts from resolution agents, simplifying effluent treatment and reducing the environmental footprint. This aligns with increasingly stringent global regulations on pharmaceutical manufacturing emissions. The high purity of the crude product also minimizes the need for extensive chromatographic purification, which is difficult to scale and solvent-intensive, further supporting sustainable large-scale production capabilities.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. Understanding these details helps stakeholders evaluate the feasibility of integrating this technology into their existing portfolios. The answers are derived directly from the experimental data and claims within the patent documentation, ensuring accuracy and relevance for technical decision-makers.
Q: How does this process improve upon traditional resolution methods?
A: Traditional methods often rely on kinetic resolution which theoretically destroys 50% of the material. This patent describes a direct enantioselective synthesis achieving ≥98% ee without the need for separation of enantiomeric salts, significantly improving atom economy.
Q: What catalysts are suitable for this asymmetric transfer hydrogenation?
A: The process utilizes chiral Ru(II) complexes, specifically Noyori-type catalysts such as Ru[(1R,2R)-p-TsNCH(C6H5)CH(C6H5)NH](η6-p-cymene). These catalysts function effectively even in the presence of trace water and do not require high-pressure hydrogen gas.
Q: Can this method be applied to different statin molecules?
A: Yes, the patent explicitly demonstrates the versatility of this route for synthesizing key intermediates for both Pitavastatin (NK-104) and Fluvastatin, accommodating different cyclic core structures like quinoline and indole derivatives.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pitavastatin Intermediate Supplier
The technological advancements detailed in CN1636004A represent a significant leap forward in the efficient production of statin intermediates. However, translating patent chemistry into commercial reality requires a partner with deep process engineering expertise and a commitment to quality. NINGBO INNO PHARMCHEM stands at the forefront of this transition, offering comprehensive CDMO services tailored to complex pharmaceutical intermediates. Our facilities are equipped to handle the specific requirements of asymmetric catalysis, including precise temperature control and advanced analytical capabilities for chiral purity assessment. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and precision. Our rigorous QC labs enforce stringent purity specifications, guaranteeing that every batch meets the high standards required for downstream API synthesis.
We invite you to collaborate with us to leverage this innovative synthesis route for your statin projects. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis that quantifies the specific economic benefits of switching to this enantioselective process for your volume requirements. We encourage you to contact our technical procurement team to request specific COA data from our pilot runs and to discuss route feasibility assessments for your specific molecular targets. Together, we can optimize your supply chain, reduce costs, and accelerate the delivery of life-saving medications to the market.