Advanced Synthesis of Rhodanine Derivatives for Potent Antineoplastic Drug Development
Advanced Synthesis of Rhodanine Derivatives for Potent Antineoplastic Drug Development
The landscape of oncology drug discovery is continuously evolving, with a significant pivot towards targeting the apoptotic pathways regulated by the Bcl-2 protein family. Patent CN102058585B introduces a groundbreaking class of rhodanine-based derivatives that exhibit potent antineoplastic activity by antagonizing these critical survival proteins. This technology represents a substantial leap forward for pharmaceutical developers seeking high-efficacy scaffolds for next-generation cancer therapeutics. The core innovation lies not only in the biological potency of the compounds but also in the robustness of the synthetic methodology employed to construct them. By leveraging a versatile rhodanine parent ring, the patent discloses a library of compounds capable of inhibiting tumor growth with remarkable specificity. 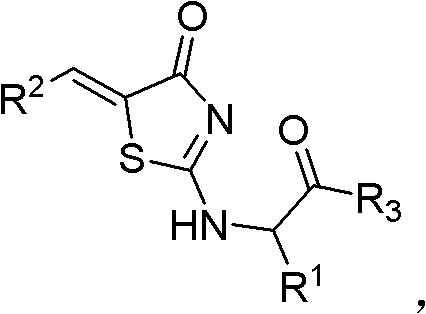 For procurement and R&D teams, this translates to a reliable pipeline of high-purity pharmaceutical intermediates that can be adapted for various oncology indications, ranging from breast carcinoma to hepatocarcinoma.
For procurement and R&D teams, this translates to a reliable pipeline of high-purity pharmaceutical intermediates that can be adapted for various oncology indications, ranging from breast carcinoma to hepatocarcinoma.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of benzyl-substituted rhodanine derivatives has been plagued by significant process inefficiencies and environmental hazards. Traditional routes often rely heavily on noble metal catalysts, such as palladium on charcoal, to facilitate hydrogenation steps required for reducing thio-groups or modifying the heterocyclic core. These methods necessitate stringent removal protocols to ensure residual metal levels meet regulatory standards for API manufacturing, adding considerable complexity and cost to the production workflow. Furthermore, alternative reduction strategies utilizing zinc in acetate mixtures generate excessive amounts of environmentally harmful zinc waste, creating disposal challenges that conflict with modern green chemistry principles. The reliance on such transition metals not only inflates the raw material costs but also introduces variability in batch-to-batch consistency due to catalyst deactivation issues. Additionally, older Mannich reaction pathways reported in literature often suffer from poor regioselectivity and require harsh conditions that can degrade sensitive functional groups, limiting the scope of substituents that can be introduced onto the rhodanine scaffold.
The Novel Approach
The methodology outlined in the patent data presents a streamlined, metal-free alternative that circumvents these historical bottlenecks through a rational four-step synthetic design. This novel approach eliminates the need for expensive hydrogenation catalysts and toxic heavy metal reductants, replacing them with standard organic transformations that are easily scalable in a multi-purpose reactor setup. The process utilizes mild conditions, such as refluxing in acetic acid for condensation and room temperature stirring for coupling reactions, which significantly reduces energy consumption and operational risks. By avoiding noble metals entirely, the downstream purification process is drastically simplified, removing the need for specialized scavenging resins or complex filtration steps associated with catalyst removal. This shift not only enhances the overall yield and purity profile of the final antineoplastic intermediate but also aligns perfectly with the increasing regulatory pressure for sustainable manufacturing practices. The result is a cost-effective, environmentally compliant route that ensures a stable supply of critical oncology building blocks for global pharmaceutical partners.
Mechanistic Insights into Rhodanine Functionalization and Coupling
The chemical elegance of this synthesis lies in its stepwise construction of the pharmacophore, beginning with a classic Knoevenagel condensation that establishes the conjugated system essential for biological activity. In the initial step, a substituted aldehyde reacts with the active methylene group of rhodanine in the presence of sodium acetate, driving the elimination of water to form the benzylidene rhodanine intermediate, designated as Compound A. This step is critical as it sets the stereochemistry and electronic properties of the R2 substituent, which interacts directly with the hydrophobic pockets of the target Bcl-2 protein. Following this, the sulfur atom at the 2-position of the thiazolidinone ring undergoes S-methylation using iodomethane and a base like diisopropylethylamine. This transformation generates Compound B, effectively activating the ring for subsequent nucleophilic attack while protecting the thione moiety from unwanted side reactions during the coupling phase. 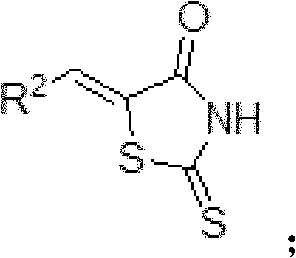
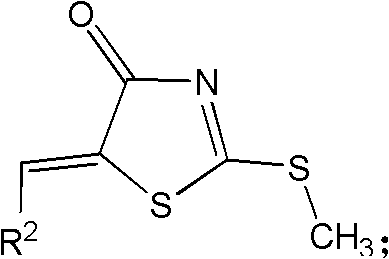 The strategic placement of the methyl group facilitates the displacement reaction in the third step, where a chiral amino acid acts as the nucleophile.
The strategic placement of the methyl group facilitates the displacement reaction in the third step, where a chiral amino acid acts as the nucleophile.
The final assembly of the molecule involves a sophisticated nucleophilic substitution followed by an amide coupling, which installs the solubilizing and targeting R3 group. In the third stage, the S-methylated intermediate reacts with an L-type natural amino acid, displacing the methyl-thio group and forming a new carbon-nitrogen bond that links the amino acid side chain (R1) to the core. This step introduces chirality into the molecule, which is often crucial for high-affinity binding to the enantioselective sites on the Bcl-2 protein family. The resulting intermediate, Compound C, possesses a free carboxylic acid handle that is primed for the final conjugation. 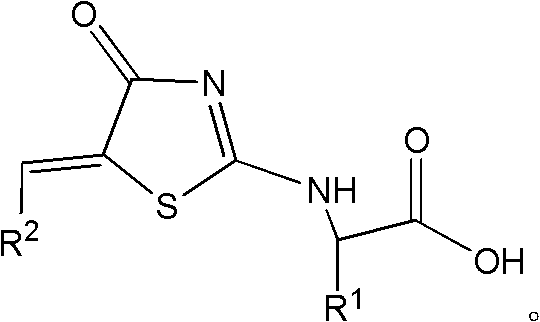 The concluding step employs standard peptide coupling reagents, specifically EDC and DMAP, to attach a sulfonamide group to the amino acid terminus. This amidation reaction is highly efficient and tolerant of various functional groups, allowing for the rapid generation of a diverse library of analogs. The mechanistic precision of this route ensures that impurities are minimized at each stage, resulting in a final product with a clean impurity profile that simplifies regulatory filing and quality control assurance for commercial scale-up of complex pharmaceutical intermediates.
The concluding step employs standard peptide coupling reagents, specifically EDC and DMAP, to attach a sulfonamide group to the amino acid terminus. This amidation reaction is highly efficient and tolerant of various functional groups, allowing for the rapid generation of a diverse library of analogs. The mechanistic precision of this route ensures that impurities are minimized at each stage, resulting in a final product with a clean impurity profile that simplifies regulatory filing and quality control assurance for commercial scale-up of complex pharmaceutical intermediates.
How to Synthesize Rhodanine Derivatives Efficiently
Executing this synthesis requires careful attention to stoichiometry and reaction monitoring to maximize yield and minimize byproduct formation, particularly during the condensation and coupling stages. The process begins by dissolving the aldehyde and rhodanine in glacial acetic acid with sodium acetate, followed by refluxing for 5 to 10 hours to ensure complete conversion to the benzylidene intermediate. Once cooled, the product is precipitated with water and filtered, providing a crude solid that can be used directly in the next step or purified if higher purity is required. The subsequent S-methylation is performed in anhydrous ethanol under inert atmosphere to prevent moisture interference, with iodomethane added dropwise to control exothermicity. After the substitution with the amino acid, the reaction mixture is concentrated and hydrolyzed with dilute hydrochloric acid to reveal the free acid functionality necessary for the final coupling. Detailed standardized synthetic steps see the guide below.
- Perform Knoevenagel condensation between substituted aldehydes and rhodanine using sodium acetate in acetic acid to form the benzylidene rhodanine intermediate.
- Execute S-methylation of the intermediate using iodomethane and diisopropylethylamine in anhydrous ethanol to activate the thiazolidinone ring.
- Conduct nucleophilic substitution with L-amino acids followed by hydrolysis to introduce the chiral side chain, forming the key amino-acid linked intermediate.
- Finalize the synthesis via EDC/DMAP mediated amide coupling with substituted sulfonamides to yield the target antineoplastic rhodanine derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible strategic benefits that extend beyond simple unit price considerations. The elimination of noble metal catalysts such as palladium removes a significant variable cost driver and mitigates the supply risk associated with fluctuating precious metal markets. Furthermore, the avoidance of heavy metal reagents like zinc drastically reduces the environmental compliance burden, lowering the costs associated with waste treatment and disposal which are often hidden expenses in traditional chemical manufacturing. The reliance on commodity chemicals like substituted benzaldehydes, amino acids, and sulfonamides ensures that the raw material supply chain is robust, diversified, and less susceptible to geopolitical disruptions compared to specialized catalytic systems. This stability allows for long-term contracting and better inventory planning, securing the continuity of supply for critical oncology programs.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by removing the need for expensive catalyst recovery systems and heavy metal scavenging resins. By utilizing standard organic reagents and ambient pressure conditions, the capital expenditure for specialized high-pressure hydrogenation equipment is rendered unnecessary. The simplified workup procedures, which primarily involve precipitation and filtration rather than complex chromatography or extraction, reduce solvent consumption and labor hours per kilogram of product. These cumulative efficiencies translate into a lower cost of goods sold (COGS), enabling more competitive pricing for the final API or intermediate without compromising on quality standards.
- Enhanced Supply Chain Reliability: The synthetic pathway relies on widely available starting materials that are produced by multiple global suppliers, reducing the risk of single-source dependency. The reaction conditions are mild and do not require cryogenic temperatures or ultra-high pressures, making the process adaptable to a wide range of manufacturing facilities with standard glass-lined or stainless steel reactors. This flexibility allows for rapid technology transfer between sites and ensures that production can be scaled up or shifted geographically with minimal downtime. Consequently, lead times for high-purity pharmaceutical intermediates can be significantly reduced, providing a buffer against market volatility and ensuring timely delivery for clinical trial materials.
- Scalability and Environmental Compliance: From a sustainability perspective, this route aligns with green chemistry metrics by minimizing waste generation and avoiding toxic reagents. The absence of heavy metal residues simplifies the environmental impact assessment and facilitates easier regulatory approval in jurisdictions with strict discharge limits. The process is inherently scalable from gram-scale laboratory synthesis to multi-ton commercial production without encountering the heat transfer or mixing limitations often seen with heterogeneous catalysis. This scalability ensures that the supply can grow in tandem with the clinical success of the drug candidate, supporting seamless transition from Phase I trials to commercial launch without the need for disruptive process redevelopment.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these rhodanine-based antineoplastic agents. These insights are derived directly from the patent specifications and are intended to clarify the feasibility and advantages of this manufacturing platform for potential partners. Understanding these details is crucial for evaluating the fit of this technology within your existing drug development portfolio.
Q: What is the primary mechanism of action for these rhodanine derivatives?
A: These derivatives function as antagonists of the anti-apoptotic Bcl-2 protein family, specifically targeting members like Bcl-2, Bcl-XL, and Mcl-1 which are overexpressed in various carcinomas including breast and prostate cancer.
Q: How does this synthesis route improve upon conventional methods?
A: Unlike traditional methods requiring noble metal catalysts like palladium or hazardous zinc reductions, this route utilizes standard organic reagents, eliminating heavy metal contamination risks and simplifying downstream purification.
Q: Are the starting materials for this process commercially viable?
A: Yes, the synthesis relies on readily available commodity chemicals such as substituted benzaldehydes, rhodanine, natural amino acids, and sulfonamides, ensuring a robust and scalable supply chain for commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Rhodanine Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a dependable partner for the supply of complex oncology intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous demands of global pharmaceutical clients. We are committed to delivering high-purity rhodanine derivatives that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our facility is designed to handle sensitive chemistries safely and efficiently, guaranteeing batch-to-batch consistency that is vital for regulatory submissions and clinical success.
We invite you to collaborate with us to leverage this advanced synthetic technology for your antineoplastic drug programs. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your project timelines and optimize your overall development budget.