Advanced Synthesis of Olaparib Impurity V for High-Purity Reference Standards
Advanced Synthesis of Olaparib Impurity V for High-Purity Reference Standards
The pharmaceutical industry's reliance on Poly (ADP-ribose) polymerase (PARP) inhibitors, such as Olaparib, has necessitated rigorous quality control measures to ensure patient safety and drug efficacy. As detailed in patent CN112279850B, the presence of specific process-related impurities, particularly the structurally complex Impurity V, poses significant challenges during the manufacturing of the active pharmaceutical ingredient (API). This patent discloses a novel, robust preparation method for Olaparib Impurity V, addressing the critical need for authentic reference substances that allow manufacturers to strictly monitor and control impurity levels. By shifting away from hazardous, low-yield historical methods, this technology offers a pathway to generate high-purity standards essential for regulatory compliance and batch release testing.
For R&D directors and quality assurance teams, the ability to source or synthesize these reference standards reliably is paramount. The patented process described herein utilizes readily available starting materials like formyl benzoic acid and employs a sequence of condensation, substitution, and dehydration reactions. This approach not only mitigates the risks associated with traditional high-temperature syntheses but also ensures that the resulting Impurity V possesses the structural integrity and purity required for precise analytical calibration. Understanding the mechanistic nuances and operational advantages of this route is vital for any organization aiming to optimize their Olaparib supply chain and maintain stringent quality specifications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
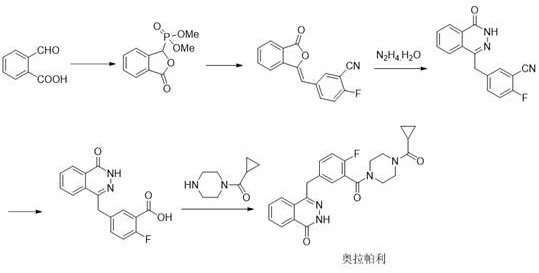
Historically, the synthesis of complex heterocyclic impurities like Compound V has been fraught with inefficiencies and safety hazards. Prior art, specifically referenced in Arch. Pharm. (1982), describes a route involving the condensation of a specific precursor with hydrazine hydrate. However, this legacy method suffers from severe selectivity issues due to the tautomerism of the starting materials, leading to a mixture of products that are difficult to separate. Furthermore, the preparation of the necessary starting compounds for these older routes often demands extreme reaction conditions, such as temperatures reaching 200°C. Such harsh environments not only present significant operational safety risks in a manufacturing setting but also result in unacceptably low yields, reported as low as 27% in some literature. These factors make the conventional production of Impurity V economically unviable and technically unreliable for generating the large quantities of high-purity material needed for comprehensive drug substance characterization.
The Novel Approach
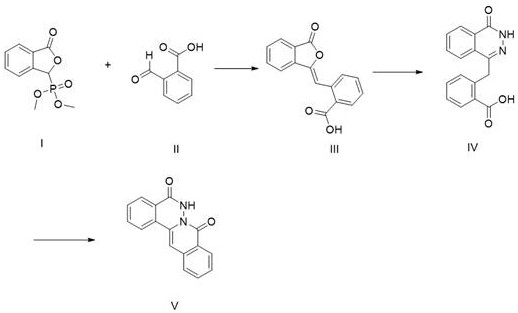
In stark contrast to the limitations of the past, the methodology outlined in patent CN112279850B introduces a streamlined, three-step synthetic strategy that prioritizes mild conditions and high selectivity. The process initiates with a condensation reaction between a phosphonate ester and formyl benzoic acid, proceeding smoothly in aqueous or organic solvents at moderate temperatures ranging from 30°C to 45°C. This is followed by a substitution reaction with hydrazine hydrate and a final cyclization step mediated by thionyl chloride. The brilliance of this approach lies in its ability to bypass the tautomerism issues of previous methods, delivering the target Impurity V with an HPLC purity exceeding 99% without the need for costly and time-consuming column chromatography purification. This represents a paradigm shift in how reference standards are manufactured, moving from artisanal, low-yield lab curiosities to scalable, industrial-grade processes.
Mechanistic Insights into the Three-Step Synthesis Strategy
The success of this novel preparation method relies on a carefully orchestrated sequence of organic transformations that maximize atom economy and minimize side reactions. The first step involves a base-catalyzed condensation, likely a Horner-Wadsworth-Emmons or similar olefination reaction, where the phosphonate species reacts with the aldehyde group of formyl benzoic acid. The use of mild inorganic bases like sodium bicarbonate at 30-45°C ensures that the sensitive carboxylic acid functionality remains intact while driving the formation of the carbon-carbon double bond in Compound III. This step is critical as it establishes the carbon skeleton of the impurity with high stereochemical control, avoiding the formation of polymeric byproducts that often plague high-temperature condensations. The subsequent isolation via pH adjustment and extraction further purifies the intermediate, setting a high baseline for the final product quality.
The final transformation involves an intramolecular cyclization facilitated by thionyl chloride (SOCl2) in the presence of a nucleophilic catalyst such as DMAP or DMF. This step effectively activates the carboxylic acid moiety, promoting nucleophilic attack by the adjacent hydrazine nitrogen to close the ring and form the phthalazinone core characteristic of Impurity V. The reaction is conducted in toluene at 75-85°C, a temperature range that provides sufficient energy for cyclization without degrading the thermally sensitive heterocyclic system. The addition of triethylamine post-reaction serves to neutralize acidic byproducts and facilitates the precipitation of the final solid. This mechanistic precision ensures that the impurity profile remains clean, with the patent reporting final purities of 99.18% to 99.5% directly after filtration and drying, demonstrating exceptional process robustness.
How to Synthesize Olaparib Impurity V Efficiently
The synthesis of Olaparib Impurity V described in this patent offers a reproducible framework for laboratories aiming to produce this critical reference standard. The process is designed to be operationally simple, utilizing common laboratory equipment such as three-necked flasks and standard heating mantles, making it accessible for both pilot-scale and commercial production environments. The protocol emphasizes the importance of temperature control during the exothermic addition of reagents and the strategic use of pH adjustments to isolate intermediates without resorting to complex purification techniques. For a detailed breakdown of the specific reagent quantities, solvent choices, and reaction times required to replicate this high-yield process, please refer to the standardized synthesis guide below.
- Condense formyl benzoic acid with a phosphonate ester in aqueous bicarbonate solution at 30-45°C to form the olefinic intermediate.
- React the olefinic intermediate with hydrazine hydrate in THF at 60-80°C to generate the hydrazide precursor.
- Perform cyclization using thionyl chloride and a catalyst like DMAP in toluene at 75-85°C, followed by base neutralization.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this patented synthesis route offers profound benefits for procurement managers and supply chain leaders tasked with sourcing high-quality pharmaceutical intermediates. The shift from hazardous, high-energy processes to mild, ambient-pressure reactions significantly reduces the operational overhead associated with manufacturing. By eliminating the need for extreme temperatures (200°C) and replacing them with温和 conditions (30-85°C), the process drastically lowers energy consumption and minimizes the risk of thermal runaway incidents. This inherent safety profile translates directly into lower insurance costs and reduced downtime, ensuring a more stable and predictable supply of critical reference materials for quality control laboratories globally.
- Cost Reduction in Manufacturing: The economic impact of this technology is driven primarily by the elimination of column chromatography, a notoriously expensive and slow purification step. Achieving >99% purity through simple crystallization and filtration means that manufacturing throughput can be significantly increased while reducing solvent waste and silica gel costs. Furthermore, the use of cheap, commodity chemicals like sodium bicarbonate and formyl benzoic acid as starting materials ensures that the raw material cost base remains low and stable, shielding the supply chain from the volatility often seen with exotic reagents.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route enhances supply chain continuity by reducing the likelihood of batch failures. Traditional methods with low yields (27%) and poor selectivity often result in inconsistent output, forcing procurement teams to seek multiple backup suppliers. In contrast, this high-selectivity process ensures consistent batch-to-batch quality, allowing for longer-term supply contracts and reduced safety stock requirements. The simplicity of the work-up procedures also means that production lead times can be shortened, enabling faster response to sudden increases in demand for Olaparib testing standards.
- Scalability and Environmental Compliance: Scalability is a key advantage, as the reaction conditions are easily transferable from gram-scale laboratory synthesis to multi-kilogram commercial production. The use of water as a solvent in the initial step and the avoidance of heavy metal catalysts align with modern green chemistry principles, simplifying wastewater treatment and environmental compliance. This eco-friendly profile not only reduces disposal costs but also aligns with the sustainability goals of major pharmaceutical companies, making the supplier of this impurity a more attractive partner in a regulated market.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Olaparib Impurity V. These answers are derived directly from the technical specifications and experimental data provided in patent CN112279850B, ensuring that stakeholders have accurate information for decision-making. Understanding these details is crucial for integrating this reference standard into your quality control workflows effectively.
Q: Why is a specific synthesis route needed for Olaparib Impurity V?
A: Impurity V is a difficult-to-separate byproduct in Olaparib manufacturing. A dedicated synthesis route allows for the production of high-purity reference standards essential for accurate HPLC quantification and quality control of the final API.
Q: How does the new method improve upon prior art synthesis?
A: Unlike previous methods requiring dangerous high temperatures (200°C) and yielding poor results (27%), this patented process operates under mild conditions (30-85°C) and achieves over 99% purity without the need for expensive column chromatography.
Q: What are the critical reagents for the final cyclization step?
A: The final conversion to Impurity V utilizes thionyl chloride as the dehydrating agent, with DMAP or DMF as a catalyst in toluene, followed by neutralization with triethylamine to ensure high selectivity and yield.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Olaparib Impurity V Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-purity reference standards play in the development and manufacturing of life-saving oncology drugs like Olaparib. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements regardless of the scale. We are committed to delivering materials that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify every batch against the highest industry standards.
We invite you to collaborate with us to optimize your supply chain for Olaparib impurities. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific needs. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our advanced synthesis capabilities can support your regulatory and commercial goals.