Advanced Resolution Technology for High-Purity Dihydropyrimidine Pharmaceutical Intermediates
The pharmaceutical industry's relentless pursuit of safer and more effective antiviral therapies has placed a premium on the development of highly specific chiral intermediates. Patent CN107674072B introduces a groundbreaking preparation method for optically pure dihydropyrimidine derivatives, specifically targeting the synthesis of potent hepatitis B inhibitors. This technology addresses a critical bottleneck in the manufacturing of nucleoside analogues by replacing inefficient racemic separation techniques with a robust, crystallization-driven resolution process. For R&D directors and procurement specialists, this patent represents a significant leap forward in process chemistry, offering a pathway to produce high-purity active pharmaceutical ingredient (API) intermediates with superior stereochemical control. The method leverages the formation of stable acid adducts, particularly L-tartaric acid complexes, to isolate the biologically active enantiomer with exceptional efficiency. By shifting away from resource-intensive chromatographic methods, this innovation not only enhances product quality but also fundamentally alters the economic landscape of producing complex heterocyclic building blocks for the global antiviral market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the production of optically active dihydropyrimidine compounds relied heavily on preparative chromatography to separate racemic mixtures into their constituent enantiomers. This traditional approach presents severe limitations for industrial-scale manufacturing, primarily due to its high operational costs and low throughput capabilities. Preparative HPLC or SFC requires specialized equipment, expensive chiral stationary phases, and large volumes of high-grade solvents, which drastically inflates the cost of goods sold (COGS). Furthermore, chromatographic processes are inherently batch-limited and difficult to scale linearly, creating bottlenecks that hinder the ability to meet commercial demand for antiviral drugs. The reliance on such methods also introduces significant supply chain risks, as the availability of chiral columns and the complexity of method validation can lead to prolonged lead times. From an environmental perspective, the excessive solvent consumption associated with chromatography generates substantial waste streams, complicating disposal and increasing the carbon footprint of the manufacturing process. These factors collectively make conventional resolution strategies unsustainable for the mass production of next-generation pharmaceutical intermediates.
The Novel Approach
In stark contrast, the methodology disclosed in CN107674072B utilizes a sophisticated diastereomeric resolution strategy that capitalizes on the differential solubility of acid adducts. Instead of forcing a separation through physical adsorption, this novel approach chemically modifies the target molecule by forming a salt or co-crystal with a chiral resolving agent, such as L-tartaric acid. This transformation converts the enantiomeric mixture into diastereomeric salts, which possess distinct physical properties, particularly solubility, allowing for separation via simple crystallization. The process involves dissolving the precursor compound in a suitable solvent system, introducing the resolving agent, and carefully controlling the temperature profile to precipitate the desired isomer selectively. This shift from chromatography to crystallization dramatically simplifies the unit operations required, enabling the use of standard stainless steel reactors and filtration equipment. The result is a process that is not only more cost-effective but also inherently more scalable, providing a reliable foundation for commercial production of high-purity dihydropyrimidine derivatives.
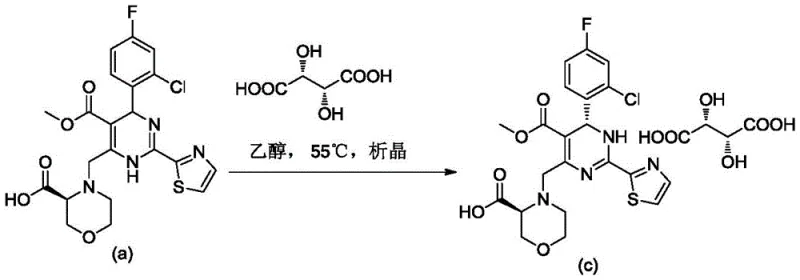
Mechanistic Insights into Chiral Resolution via Acid Adduct Formation
The core mechanism driving this purification technology is the thermodynamic stabilization of the desired enantiomer through specific intermolecular interactions with the chiral acid. When the dihydropyrimidine precursor, which contains basic nitrogen centers within its heterocyclic ring system, interacts with L-tartaric acid, it forms a stable ionic lattice structure. The patent data indicates that this interaction is highly stereoselective, favoring the crystallization of the (S)-configured isomer over its (R)-counterpart. The crystallographic analysis provided in the patent confirms the formation of a well-defined monoclinic crystal system (space group P21), where the molecular packing is optimized for the target diastereomer. This structural integrity is crucial for excluding impurities and the unwanted enantiomer from the growing crystal lattice, thereby achieving optical purities exceeding 96% in a single step. The process is further refined by optimizing the solvent environment; polar protic solvents like ethanol facilitate the ionization necessary for salt formation while maintaining sufficient solubility of the impurities in the mother liquor. By manipulating parameters such as cooling rates and stirring times, manufacturers can control the nucleation and growth kinetics to maximize yield without compromising chiral integrity.
Impurity control is another critical aspect of this mechanistic framework. The presence of structurally related byproducts or unreacted starting materials can interfere with crystal growth, potentially leading to occlusion or surface adsorption of impurities. The patented method mitigates this risk through a multi-stage purification protocol that may include a preliminary solvent wash to remove non-ionic contaminants before the acid addition step. Additionally, the choice of L-tartaric acid is strategic; it is a naturally occurring, non-toxic, and inexpensive chiral pool material that forms salts with high lattice energy, ensuring stability during downstream processing. The robustness of this acid-adduct formation allows for flexibility in the substitution patterns of the dihydropyrimidine core, accommodating various halogenated phenyl groups and heterocyclic substituents without losing resolution efficiency. This versatility makes the technology applicable to a broad library of analogues, supporting the rapid development of structure-activity relationship (SAR) campaigns in drug discovery.
How to Synthesize Optically Pure Dihydropyrimidine Efficiently
The synthesis of these high-value intermediates follows a streamlined workflow designed for reproducibility and safety. The process begins with the dissolution of the racemic or diastereomeric dihydropyrimidine precursor in a heated alcohol solvent, ensuring a homogeneous solution free of particulates. Once the temperature is stabilized, typically around 55°C, the chiral resolving agent is introduced in a stoichiometric ratio optimized to drive the equilibrium towards the desired salt. The mixture is then subjected to a controlled cooling ramp, often descending to ambient temperature or lower, which triggers the selective precipitation of the target enantiomer complex. This crystallization phase is critical and requires precise agitation to prevent agglomeration and ensure uniform crystal size distribution. Following the isolation of the solid via filtration, a washing step with cold solvent removes residual mother liquor, and a final drying stage under vacuum yields the purified product ready for subsequent coupling reactions. Detailed standardized synthetic steps see the guide below.
- Dissolve the dihydropyrimidine precursor in a suitable alcohol solvent such as ethanol and heat the mixture to approximately 55°C to ensure complete solubilization.
- Add L-tartaric acid to the solution maintaining the temperature, then slowly cool the mixture to 25°C while stirring continuously to induce selective crystallization of the desired enantiomer complex.
- Filter the precipitated solid, wash with cold solvent to remove impurities, and dry under vacuum at elevated temperatures to obtain the high-purity optical isomer.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this resolution technology translates into tangible strategic advantages that extend beyond mere technical feasibility. The elimination of preparative chromatography removes a major cost driver from the manufacturing budget, significantly reducing the expenditure on specialized consumables and high-purity solvents. This cost reduction in pharmaceutical intermediate manufacturing is achieved not by cutting corners on quality, but by engineering a more efficient process flow that relies on commodity chemicals and standard equipment. The simplified workflow also shortens the overall production cycle time, allowing for faster turnover of batches and improved responsiveness to market demand fluctuations. Furthermore, the use of common solvents like ethanol and ethyl acetate simplifies solvent recovery and recycling protocols, contributing to a more sustainable and environmentally compliant operation. These factors collectively enhance the reliability of the supply chain, reducing the risk of production delays caused by equipment failures or raw material shortages associated with more complex separation technologies.
- Cost Reduction in Manufacturing: The transition from chromatographic separation to crystallization-based resolution eliminates the need for expensive chiral columns and the high volume of mobile phase solvents typically required for HPLC purification. This fundamental shift in unit operations drastically lowers the variable costs per kilogram of product. Additionally, the resolving agent, L-tartaric acid, is a bulk commodity chemical with a stable global supply, ensuring that raw material costs remain predictable and low. The ability to recover and recycle the mother liquor for further processing or solvent reclamation further amplifies these savings, creating a lean manufacturing model that maximizes resource utilization while minimizing waste disposal fees.
- Enhanced Supply Chain Reliability: Scalability is a primary concern for any commercial API intermediate, and this technology offers a clear path from gram-scale laboratory synthesis to multi-ton commercial production. Because the process utilizes standard stirred-tank reactors and filtration units, it does not require custom-engineered equipment that could introduce long lead times for installation and qualification. This ease of scale-up ensures that supply can be ramped up quickly to meet clinical trial demands or commercial launch volumes without the need for extensive process re-validation. The robustness of the crystallization process also means that batch-to-batch variability is minimized, providing consistent quality that simplifies regulatory filings and reduces the risk of supply disruptions due to out-of-specification results.
- Scalability and Environmental Compliance: The environmental profile of this manufacturing route is significantly improved compared to traditional methods. By avoiding the massive solvent consumption inherent in chromatography, the process reduces the facility's volatile organic compound (VOC) emissions and liquid waste generation. The solvents employed are generally classified as Class 3 or low-risk solvents under ICH guidelines, facilitating easier handling and disposal. Moreover, the high yield and purity achieved in fewer steps mean less energy is consumed per unit of product, aligning with corporate sustainability goals. This greener manufacturing footprint not only satisfies regulatory requirements but also enhances the brand reputation of the supplier as a responsible partner in the pharmaceutical value chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this chiral resolution technology. These insights are derived directly from the experimental data and claims within the patent documentation, providing a factual basis for evaluating the process viability. Understanding these details is essential for technical teams assessing the fit of this intermediate within their existing synthetic routes and for commercial teams negotiating supply agreements. The answers reflect the proven capabilities of the method as demonstrated in the provided examples.
Q: Why is optical purity critical for dihydropyrimidine antiviral intermediates?
A: Pharmacokinetic studies indicate significant differences in metabolic stability and activity between stereoisomers of dihydropyrimidine compounds. High optical purity ensures consistent therapeutic efficacy and reduces the risk of toxicity associated with inactive or harmful enantiomers in hepatitis B treatments.
Q: How does this resolution method improve manufacturing scalability compared to chromatography?
A: Traditional preparative chromatography requires expensive equipment and has limited throughput, making it unsuitable for multi-kilogram production. This crystallization-based resolution utilizes standard reactor vessels and common solvents, allowing for seamless scale-up from laboratory to commercial tonnage without specialized separation columns.
Q: What solvents are compatible with this chiral resolution process?
A: The process demonstrates robust compatibility with a wide range of green and industrial solvents including ethanol, methanol, isopropanol, and ethyl acetate. This flexibility allows manufacturers to optimize solvent recovery systems and reduce environmental impact while maintaining high yield and purity specifications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Dihydropyrimidine Derivatives Supplier
NINGBO INNO PHARMCHEM stands at the forefront of implementing advanced chiral resolution technologies to deliver high-quality pharmaceutical intermediates to the global market. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless and efficient. We understand that the consistency of optical purity is non-negotiable for antiviral drug candidates, which is why we employ stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art chiral HPLC and X-ray diffraction capabilities. Our commitment to quality assurance guarantees that every batch of dihydropyrimidine derivative meets the exacting standards required for GMP manufacturing, providing our partners with the confidence needed to advance their drug development programs.
We invite potential partners to engage with our technical procurement team to discuss how this patented resolution method can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can quantify the potential economic benefits of switching to this crystallization-based process for your specific project needs. We encourage you to contact us to obtain specific COA data for our current inventory and to request detailed route feasibility assessments tailored to your target molecules. Together, we can accelerate the delivery of life-saving antiviral therapies to patients worldwide through superior chemistry and reliable supply chain execution.