Advanced Synthesis of 2-Bromo-6-Fluoroaniline for High-Purity Pharmaceutical Intermediate Manufacturing
Introduction to Next-Generation Synthetic Pathways
The pharmaceutical industry constantly seeks robust, safe, and cost-effective routes for critical intermediates, particularly those serving antiviral applications such as Letermovir synthesis. A groundbreaking development in this sector is detailed in patent CN115784896A, which discloses a superior preparation method for 2-bromo-6-fluoroaniline. This compound serves as a pivotal building block in the manufacture of CMV DNA terminase complex inhibitors, essential for preventing Cytomegalovirus infections in immunocompromised patients undergoing hematopoietic stem cell transplantation. The disclosed technology represents a paradigm shift from hazardous, low-yield legacy processes to a sophisticated, multi-step protection strategy that ensures both operational safety and exceptional chemical purity. By ingeniously utilizing sulfonamide groups as temporary blocking agents, this method effectively circumvents the formation of stubborn para-position impurities that have long plagued direct bromination attempts. For global procurement teams and R&D directors, understanding this technological leap is crucial for securing a reliable pharmaceutical intermediate supplier capable of delivering consistent quality without the supply chain disruptions associated with controlled explosive precursors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-bromo-6-fluoroaniline has been fraught with significant safety and efficiency challenges that hinder large-scale commercial viability. The first conventional pathway, often cited in literature, relies on 2-bromo-6-fluorobenzoic acid as a starting material, which undergoes acidification with concentrated sulfuric acid followed by a reaction with sodium azide and quenching with ammonia water. This route is fundamentally flawed for modern industrial standards because sodium azide is a notorious explosive hazard, requiring extreme caution and specialized containment facilities that drastically inflate capital expenditure and operational risk. Furthermore, the starting material, 2-bromo-6-fluorobenzoic acid, is not readily available in bulk quantities and commands a high market price, creating a bottleneck for cost reduction in API manufacturing.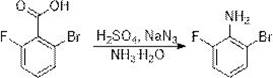
The second conventional approach attempts to bypass the expensive acid precursor by starting directly with o-fluoroaniline and employing various brominating agents such as elemental bromine or hydrobromic acid. However, this direct electrophilic aromatic substitution suffers from inherent selectivity issues. The amino group is a strong activator, directing incoming electrophiles to both ortho and para positions. Consequently, this method generates a complex mixture of isomers, specifically the unwanted para-brominated byproducts, which are structurally similar to the target molecule and notoriously difficult to separate via standard crystallization or distillation. The resulting low yield and arduous purification processes render this route economically unviable for high-volume production, failing to meet the stringent purity specifications required by regulatory bodies for active pharmaceutical ingredients.
The Novel Approach
In stark contrast to these legacy methods, the technology outlined in CN115784896A introduces a clever "protect-block-react-deprotect" strategy that resolves both safety and selectivity dilemmas simultaneously. Instead of risking explosive azides or battling poor regioselectivity, this novel route begins with the inexpensive and widely available o-fluoroaniline. The process initiates by protecting the sensitive amino group, followed by a critical sulfonylation step that installs a bulky sulfonamide moiety specifically at the para-position. This sulfonamide group acts as a steric and electronic shield, effectively blocking the para-site from subsequent electrophilic attack. When the bromination step is subsequently performed, the bromine atom is forced exclusively into the remaining open ortho-position, guaranteeing high regioselectivity. Finally, the protecting groups are cleaved under acidic conditions to reveal the pristine 2-bromo-6-fluoroaniline. This approach not only eliminates the need for hazardous sodium azide but also transforms a difficult separation problem into a straightforward synthetic sequence, offering a clear path toward commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Regioselective Sulfonylation and Bromination
To fully appreciate the technical sophistication of this synthesis, one must examine the mechanistic role of the sulfonamide blocking group in controlling the reaction trajectory. The process begins with the acylation of o-fluoroaniline to form Intermediate 1, which moderates the nucleophilicity of the nitrogen atom. Subsequently, the introduction of chlorosulfonic acid generates a sulfonyl chloride species in situ, which reacts preferentially at the para-position relative to the amino group due to steric and electronic factors favored by the existing substituents. The conversion of this sulfonyl chloride into a sulfonamide (Intermediate 2) using ammonia creates a robust electron-withdrawing group that deactivates the aromatic ring towards further electrophilic substitution at that specific site. This deactivation is the key to the method's success; when the brominating agent (preferably a hydrogen bromide/hydrogen peroxide system) is introduced, the para-position is chemically inert, leaving the ortho-position as the only viable site for reaction. This precise control over molecular architecture ensures that the resulting Intermediate 3 possesses the exact substitution pattern required, minimizing the formation of isomeric impurities that would otherwise compromise the final drug substance's safety profile.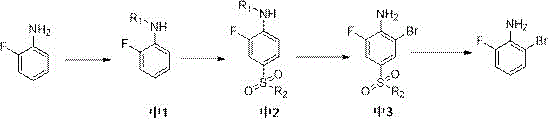
Furthermore, the final deprotection step is engineered for maximum efficiency and minimal side reactions. The removal of both the amino protecting group (e.g., acetyl) and the para-sulfonamide blocking group is achieved in a single pot using concentrated sulfuric acid at elevated temperatures (150-180°C). This harsh acidic environment hydrolyzes the amide and sulfonamide bonds rapidly, regenerating the free amine and releasing the sulfonic acid byproduct, which remains soluble in the aqueous phase. The target 2-bromo-6-fluoroaniline, being an organic base, can then be easily extracted into an organic solvent like dichloromethane. This streamlined workup procedure avoids the need for multiple isolation steps between deprotection events, significantly reducing solvent consumption and processing time. The result is a final product with purity levels exceeding 99.6% and single impurity content below 0.1%, demonstrating the method's capability to produce high-purity pharmaceutical intermediates that meet the rigorous demands of global regulatory agencies without the need for extensive chromatographic purification.
How to Synthesize 2-Bromo-6-Fluoroaniline Efficiently
The practical implementation of this synthesis involves four distinct operational stages that balance reaction kinetics with safety protocols. The process starts with the protection of o-fluoroaniline in a solvent like dichloromethane at low temperatures (0-20°C) to prevent exothermic runaway, followed by the critical sulfonylation step where temperature control is vital to manage the evolution of gases and ensure complete conversion to the sulfonamide. The subsequent bromination utilizes an oxidative system that is safer than handling liquid bromine directly, and the final hydrolysis requires corrosion-resistant equipment capable of withstanding hot concentrated sulfuric acid. For a detailed breakdown of the specific molar ratios, temperature profiles, and workup procedures validated in the patent examples, please refer to the standardized protocol below.
- Protect the amino group of o-fluoroaniline using acetyl chloride or similar agents to form Intermediate 1.
- Perform sulfonylation on Intermediate 1 using chlorosulfonic acid followed by amidation to block the para-position, yielding Intermediate 2.
- Conduct bromination on Intermediate 2 using a hydrogen bromide/hydrogen peroxide system to obtain Intermediate 3 with high regioselectivity.
- Remove the sulfonamide and amino protecting groups from Intermediate 3 under acidic conditions (e.g., sulfuric acid at 160°C) to isolate the target product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers transformative benefits that extend far beyond simple chemical yield improvements. By shifting away from the traditional azide-based Route 1, manufacturers can eliminate the substantial logistical and insurance costs associated with transporting and storing explosive sodium azide. This change alone simplifies the regulatory compliance landscape, allowing for smoother audits and reduced liability exposure. Moreover, the reliance on commodity chemicals like o-fluoroaniline, chlorosulfonic acid, and hydrogen peroxide ensures that raw material sourcing is resilient against market volatility. Unlike specialized fluorinated benzoic acids which may have limited suppliers, these bulk chemicals are produced globally in massive quantities, guaranteeing reducing lead time for high-purity pharmaceutical intermediates and preventing production stoppages due to raw material shortages. The robustness of the supply chain is further enhanced by the process tolerance, which accommodates slight variations in reagent quality without compromising the final product specification.
- Cost Reduction in Manufacturing: The economic advantages of this method are driven by the elimination of expensive purification steps and the use of low-cost starting materials. Traditional routes often require multiple recrystallizations or column chromatography to remove para-isomers, which consumes vast amounts of solvent and labor. By preventing the formation of these isomers at the source through the blocking group strategy, the new process drastically reduces downstream processing costs. Additionally, the avoidance of precious metal catalysts or exotic reagents means that the cost of goods sold (COGS) is significantly lowered, allowing for more competitive pricing strategies in the generic drug market while maintaining healthy margins for the manufacturer.
- Enhanced Supply Chain Reliability: Supply continuity is a critical metric for any pharmaceutical buyer, and this synthesis route excels in reliability by removing dependency on hazardous, controlled substances. The logistical complexity of shipping Class 1 explosives like sodium azide often leads to delays and requires specialized freight forwarders. By replacing this with standard industrial acids and oxidants, the supply chain becomes more agile and less prone to regulatory bottlenecks. Furthermore, the patent demonstrates successful scaling from gram-scale laboratory experiments to 250 kg pilot batches, proving that the chemistry holds up under production conditions. This scalability assures buyers that their suppliers can ramp up volume quickly to meet sudden spikes in demand for antiviral medications without encountering unforeseen technical barriers.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this process aligns perfectly with modern green chemistry principles. The replacement of explosive azides reduces the risk of catastrophic accidents, protecting both personnel and facility infrastructure. The solvent systems employed, primarily dichloromethane, are well-understood and can be efficiently recovered and recycled using standard distillation units, minimizing waste discharge. The high atom economy of the bromination step, coupled with the high yield of the final deprotection, ensures that waste generation per kilogram of product is minimized. This reduced environmental footprint not only lowers waste disposal costs but also enhances the corporate sustainability profile of the manufacturing entity, a factor increasingly weighted in vendor selection criteria by major multinational pharmaceutical corporations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of 2-bromo-6-fluoroaniline using this advanced sulfonylation methodology. These answers are derived directly from the experimental data and technical disclosures found in the patent literature, providing a transparent view of the process capabilities. Understanding these details helps stakeholders make informed decisions regarding vendor qualification and process integration.
Q: Why is the traditional azide route for 2-bromo-6-fluoroaniline considered unsafe for industrial scale?
A: The conventional Route 1 utilizes sodium azide (NaN3), which is a highly explosive and toxic reagent. Handling large quantities of sodium azide poses severe safety risks and requires specialized, costly infrastructure for explosion containment, making it unsuitable for standard commercial production facilities.
Q: How does the new sulfonylation method improve product purity compared to direct bromination?
A: Direct bromination of o-fluoroaniline often suffers from poor regioselectivity, leading to significant para-brominated impurities that are difficult to separate. The novel method employs a sulfonamide blocking group at the para-position, physically preventing substitution there and forcing the bromine atom exclusively to the desired ortho-position, thereby achieving purities exceeding 99%.
Q: Is this synthesis route scalable for metric-ton production?
A: Yes, the patent explicitly demonstrates scalability with Example Two detailing a 250 kg batch size run. The process utilizes common industrial solvents like dichloromethane and standard reagents like chlorosulfonic acid, avoiding exotic catalysts, which facilitates straightforward scale-up from pilot plant to commercial manufacturing volumes.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Bromo-6-Fluoroaniline Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to safer, more efficient synthetic routes is essential for the future of pharmaceutical manufacturing. Our team of expert process chemists has thoroughly analyzed the technology described in CN115784896A and is fully prepared to implement this advanced sulfonylation strategy at commercial scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with unwavering consistency. Our state-of-the-art facilities are equipped with corrosion-resistant reactors suitable for high-temperature acid hydrolysis and rigorous QC labs capable of verifying stringent purity specifications, including the detection of trace isomeric impurities down to 0.1%. We are committed to delivering a product that not only meets but exceeds the quality expectations of the global antiviral market.
We invite you to collaborate with us to optimize your supply chain for Letermovir and related antiviral intermediates. By leveraging our technical expertise and this novel synthetic route, we can offer you a Customized Cost-Saving Analysis that quantifies the potential reductions in both raw material and processing expenses. We encourage you to contact our technical procurement team today to request specific COA data from our recent pilot runs and to discuss route feasibility assessments tailored to your specific project timelines. Let us partner with you to secure a sustainable, high-quality supply of 2-bromo-6-fluoroaniline that drives your drug development forward.