Advanced Manufacturing of Faropenem Medoxomil: Technical Upgrades and Commercial Scalability
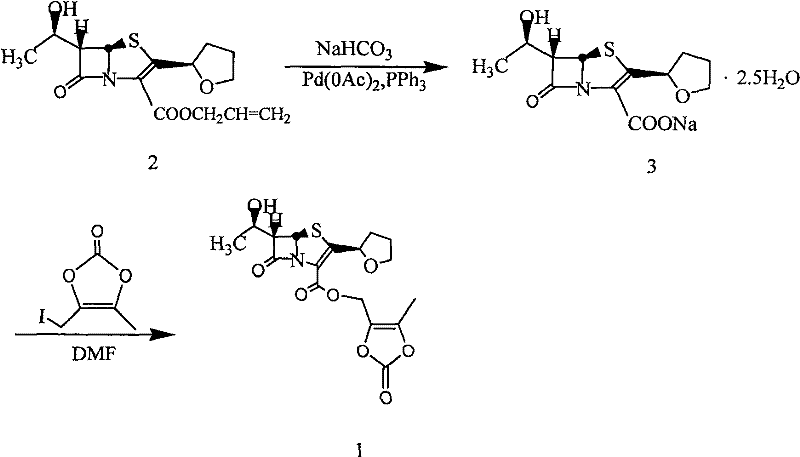
The pharmaceutical landscape for oral carbapenems is continuously evolving, with patent CN101235044B representing a pivotal advancement in the synthesis of faropenem medoxomil. This specific intellectual property outlines a robust preparation method that addresses critical bottlenecks found in earlier generations of synthetic routes. As a first-generation orally administered penem antibacterial prodrug, faropenem medoxomil requires precise chemical engineering to ensure bioavailability and stability upon ingestion. The disclosed technology leverages a sophisticated interplay of phase transfer catalysis and iodide promotion to facilitate the esterification of faropenem under remarkably mild conditions. This innovation is not merely a laboratory curiosity but a substantial leap forward for industrial manufacturers seeking to optimize their production lines for high-value antibiotic intermediates. By shifting away from harsh conditions and complex multi-step sequences, this methodology offers a pathway to enhanced operational efficiency and superior product consistency.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of faropenem medoxomil has been plagued by inefficient methodologies that rely on cumbersome reaction sequences and expensive reagents. Prior art, such as the methods described in US Patent 5885981, often necessitates the use of allyl faropenem as a starting material, requiring a palladium-catalyzed deallylation step that introduces significant complexity and cost. These traditional routes frequently suffer from low overall yields, often hovering around 39% or even lower in practical industrial settings, primarily due to the degradation of the sensitive beta-lactam ring during harsh processing conditions. Furthermore, the reliance on column chromatography for purification is a major impediment to scalability, as it is time-consuming, solvent-intensive, and difficult to implement in large-scale continuous manufacturing environments. The use of expensive reagents like tetrabutylammonium fluoride for deprotection further exacerbates the cost structure, making the final active pharmaceutical ingredient economically challenging to produce competitively.
The Novel Approach
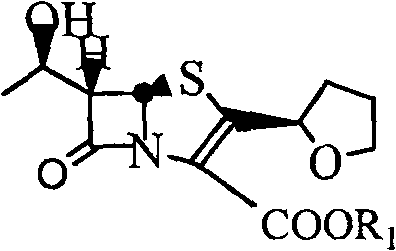
In stark contrast, the methodology detailed in CN101235044B introduces a streamlined direct esterification strategy that bypasses the need for protective group manipulation and transition metal catalysis. This novel approach utilizes faropenem or its inorganic salts directly reacting with 4-halomethyl-5-methyl-1,3-dioxol-2-one in the presence of a phase transfer catalyst and an iodide-containing promoter. The reaction proceeds efficiently in aprotic polar solvents at temperatures ranging from 0°C to 20°C, which drastically reduces the thermal stress on the delicate molecular structure. By eliminating the deallylation step and the subsequent complex purification requirements, this method simplifies the entire workflow into a more manageable and controllable process. The result is a significant enhancement in both reaction yield and product purity, achieved through a mechanism that favors the desired nucleophilic substitution while suppressing competing degradation pathways.
Mechanistic Insights into Phase Transfer Catalyzed Esterification
The core of this technological breakthrough lies in the synergistic action of the phase transfer catalyst and the iodide reaction accelerator within the alkaline reaction medium. The phase transfer catalyst, such as 18-crown-6 or polyethylene glycol, facilitates the transport of the faropenem anion from the aqueous or solid phase into the organic solvent phase where the electrophilic halomethyl dioxolone resides. This solubilization effect dramatically increases the effective concentration of the reactive species, thereby accelerating the reaction kinetics without the need for elevated temperatures that could compromise the stability of the beta-lactam core. Simultaneously, the iodide ion acts as a potent nucleophilic catalyst, potentially forming a more reactive intermediate in situ that undergoes rapid substitution with the faropenem carboxylate. This dual-catalyst system ensures that the reaction reaches completion within a short timeframe, typically between 4 to 8 hours, while maintaining a high degree of stereochemical integrity essential for the biological activity of the final drug substance.
Impurity control is another critical aspect where this mechanistic design excels, particularly concerning the preservation of the stereocenters at the C5 and C6 positions of the penem nucleus. Traditional methods often struggle with epimerization or hydrolysis of the beta-lactam ring due to prolonged exposure to basic conditions or heat. However, by maintaining the pH within a narrow range of 7.5 to 8.5 and keeping the reaction temperature low, the new process effectively mitigates these degradation risks. The use of specific alkali metal carbonates or bicarbonates provides a buffered environment that is sufficiently basic to generate the nucleophilic carboxylate but mild enough to prevent ring opening. Furthermore, the workup procedure involving the addition of water induces crystallization directly, which serves as an additional purification step by excluding soluble impurities and unreacted starting materials from the crystal lattice, thus ensuring a high-purity profile without the need for chromatographic separation.
How to Synthesize Faropenem Medoxomil Efficiently
Implementing this synthesis route requires careful attention to solvent selection and reagent stoichiometry to maximize the benefits of the phase transfer system. The process begins with the dissolution of the faropenem substrate in a solvent like 1,3-dimethylpropylene urea or a mixture of tetrahydrofuran and acetone, followed by the precise addition of the base to generate the reactive salt in situ. Once the reaction mixture is cooled to the optimal temperature range, the halomethylating agent is added dropwise to control the exotherm and prevent local overheating. The detailed standardized synthesis steps see the guide below for exact parameters regarding stirring rates, addition times, and crystallization protocols that have been validated for reproducibility.
- Dissolve faropenem or its inorganic salt in an aprotic polar solvent such as 1,3-dimethylpropylene urea or tetrahydrofuran under controlled alkaline conditions.
- Add a phase transfer catalyst like 18-crown-6 or polyethylene glycol along with an iodide-containing reaction accelerator such as potassium iodide.
- React with 4-halomethyl-5-methyl-1,3-dioxol-2-one at temperatures between 0°C to 20°C, followed by aqueous workup and recrystallization to isolate the pure ester.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic procurement perspective, the adoption of this patented synthesis method offers profound advantages in terms of cost structure and supply chain resilience. The elimination of palladium catalysts and expensive fluorinating agents removes significant variable costs associated with raw material acquisition and hazardous waste disposal. Moreover, the simplification of the downstream processing, specifically the replacement of column chromatography with crystallization, drastically reduces solvent consumption and processing time, leading to substantial cost savings in manufacturing operations. This efficiency gain translates directly into a more competitive pricing model for the final intermediate, allowing pharmaceutical companies to optimize their bill of materials for generic or branded antibiotic formulations.
- Cost Reduction in Manufacturing: The removal of transition metal catalysts and chromatographic purification steps significantly lowers the operational expenditure per kilogram of produced material. By avoiding the use of precious metals like palladium, manufacturers eliminate the need for costly metal scavenging processes and reduce the risk of heavy metal contamination in the final product. The simplified workup procedure also reduces labor hours and energy consumption associated with solvent recovery and distillation, contributing to a leaner and more cost-effective production cycle that enhances overall profit margins.
- Enhanced Supply Chain Reliability: The reagents required for this novel method, such as alkali metal carbonates and common phase transfer catalysts, are widely available commodities with stable supply chains. This contrasts sharply with older methods that rely on specialized or proprietary reagents which may be subject to supply disruptions or price volatility. By utilizing readily accessible raw materials, manufacturers can secure long-term supply agreements and reduce the risk of production stoppages, ensuring a consistent flow of high-purity faropenem medoxomil to meet market demand without interruption.
- Scalability and Environmental Compliance: The inherent design of this process favors large-scale production due to its reliance on crystallization rather than chromatography, which is notoriously difficult to scale. The reduced solvent usage and absence of toxic heavy metals simplify waste treatment protocols, making it easier to comply with increasingly stringent environmental regulations. This scalability ensures that production volumes can be ramped up quickly to meet surges in demand, while the greener chemical profile supports corporate sustainability goals and reduces the environmental footprint of the manufacturing facility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the patent specifications and are intended to clarify the operational benefits and feasibility of adopting this route for commercial production. Understanding these details is crucial for technical teams evaluating the potential for process integration and for procurement officers assessing the long-term value proposition of this manufacturing method.
Q: How does the new method improve upon conventional palladium-catalyzed routes?
A: The novel approach eliminates the need for expensive palladium catalysts and complex deprotection steps, significantly simplifying the workflow and reducing heavy metal contamination risks.
Q: What are the key advantages regarding product purity and yield?
A: By utilizing mild reaction temperatures and specific iodide promoters, the process minimizes side reactions, achieving yields exceeding 80% with high HPLC purity suitable for pharmaceutical standards.
Q: Is this synthesis route scalable for industrial production?
A: Yes, the method avoids column chromatography in favor of crystallization, making it highly amenable to large-scale manufacturing with consistent quality and reduced operational costs.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Faropenem Medoxomil Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic routes in securing the supply of essential antibiotic intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patent CN101235044B are fully realized in an industrial setting. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of faropenem medoxomil meets the highest pharmacopeial standards. Our commitment to technical excellence allows us to navigate the complexities of beta-lactam chemistry with precision, delivering products that support the development of safe and effective oral antimicrobial therapies.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific supply chain requirements. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic impact of switching to this methodology. We encourage potential partners to contact us for specific COA data and route feasibility assessments, enabling you to make informed decisions that drive efficiency and reliability in your pharmaceutical manufacturing operations.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →