Advanced Synthetic Route for Lumefantrine Impurity 3: Ensuring Quality Control in Antimalarial Manufacturing
Advanced Synthetic Route for Lumefantrine Impurity 3: Ensuring Quality Control in Antimalarial Manufacturing
The global fight against malaria relies heavily on Artemisinin-based Combination Therapies (ACTs), with Lumefantrine serving as a critical partner drug due to its efficacy against multidrug-resistant Plasmodium falciparum. As regulatory scrutiny intensifies, particularly from agencies like the US FDA and the European Medicines Agency, the requirement for comprehensive impurity profiling has become non-negotiable for market approval. Patent CN116023387A addresses a significant gap in this supply chain by disclosing a novel, five-step preparation method for Lumefantrine Impurity 3, a key degradation product listed in the European Pharmacopoeia. Previously, the lack of a defined synthetic route meant that obtaining high-purity reference standards was difficult, often relying on inefficient isolation from crude drug substances. This new methodology not only secures the supply of essential quality control materials but also demonstrates a robust chemical pathway that can be adapted for the commercial scale-up of complex pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the disclosure of this patent, the chemical community faced a significant challenge regarding the availability of Lumefantrine Impurity 3. The background section of the patent explicitly states that no relevant literature reports existed for the synthesis of this specific compound. In the absence of a dedicated synthetic route, quality control laboratories were forced to rely on isolating trace amounts of the impurity from manufactured Lumefantrine batches, a process that is inherently inefficient, costly, and yields material of inconsistent purity. Furthermore, without a known structure-activity relationship or a reliable synthesis, validating analytical methods for this impurity was fraught with uncertainty. This scarcity created a bottleneck for generic drug manufacturers seeking to prove bioequivalence and safety, as they could not accurately quantify this specific degradant in their final formulations, potentially risking regulatory rejection or delayed market entry for life-saving antimalarial medications.
The Novel Approach
The patented innovation introduces a logical and convergent synthetic strategy starting from the inexpensive and readily available raw material, 2,7-dichlorofluorene. Unlike previous undefined methods, this route clearly delineates five distinct chemical transformations: Friedel-Crafts acylation, nucleophilic addition, cyclization, hydroboration reduction, and final condensation. This step-wise approach allows for precise control over the molecular architecture, ensuring that the resulting Impurity 3 matches the structural requirements of the Pharmacopoeia with high fidelity. By building the molecule from simple precursors rather than isolating it, manufacturers can produce gram-to-kilogram quantities of the reference standard on demand. This shift from isolation to total synthesis represents a paradigm shift in how critical impurity standards are sourced, offering a reliable pharmaceutical intermediate supplier pathway that guarantees both structural integrity and batch-to-batch consistency.
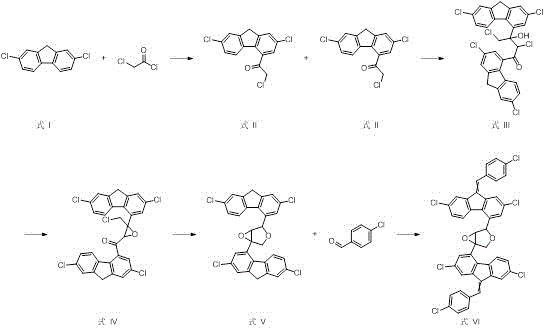
Mechanistic Insights into the Five-Step Synthetic Cascade
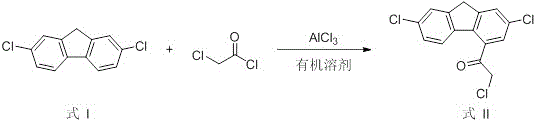
The core of this synthetic achievement lies in the strategic construction of the carbon skeleton through a series of well-understood yet precisely controlled organic reactions. The process initiates with a Friedel-Crafts acylation, where 2,7-dichlorofluorene reacts with chloroacetyl chloride in the presence of aluminum trichloride. This electrophilic aromatic substitution is critical as it installs the reactive chloroacetyl handle necessary for subsequent dimerization. The reaction is conducted at low temperatures, typically between -5°C and 30°C, to prevent poly-acylation and ensure regioselectivity at the desired position on the fluorene ring. Following this, the route employs a powerful nucleophilic addition using Lithium Diisopropylamide (LDA) at cryogenic temperatures (-78°C to -40°C). This step generates a reactive enolate species that attacks a second molecule of the acylated intermediate, effectively doubling the molecular weight and establishing the central bis-fluorene linkage that characterizes the impurity's core structure.
Following the dimerization, the synthesis proceeds through a sophisticated cyclization sequence that constructs the complex ether bridge found in the final impurity. Treatment with sodium hydride facilitates an intramolecular nucleophilic attack, closing a ring to form a cyclic ether intermediate. Subsequent reduction with sodium borohydride modifies the oxidation state of the carbonyl group, preparing the molecule for the final condensation step. The process concludes with a base-catalyzed condensation with p-chlorobenzaldehyde, which installs the final aryl-vinyl moiety. From an impurity control perspective, this route is advantageous because each intermediate (Formulas II through V) can be purified via standard techniques like column chromatography or recrystallization before proceeding to the next step. This modular purification strategy prevents the carryover of side products, ensuring that the final Lumefantrine Impurity 3 is obtained with the high purity specifications required for analytical reference standards.
How to Synthesize Lumefantrine Impurity 3 Efficiently
The practical execution of this synthesis requires careful attention to reaction conditions, particularly temperature control and stoichiometry, to maximize yield and minimize byproduct formation. The patent outlines specific solvent systems for each step, such as using dichloromethane for the initial acylation and tetrahydrofuran for the low-temperature lithiation, ensuring optimal solubility and reaction kinetics. Operators must strictly adhere to the molar ratios specified, such as using 1.2 equivalents of chloroacetyl chloride and 1.5 equivalents of Lewis acid in the first step, to drive the reaction to completion without excessive waste. The detailed standardized synthesis steps below provide a roadmap for laboratory technicians to reproduce this high-value intermediate reliably.
- Perform Friedel-Crafts acylation on 2,7-dichlorofluorene using chloroacetyl chloride and aluminum trichloride to form the mono-acylated intermediate.
- Execute a nucleophilic addition dimerization using LDA at low temperatures to couple two acylated units, forming the bis-fluorene ketone structure.
- Conduct intramolecular cyclization using sodium hydride, followed by borohydride reduction and base-catalyzed ring closure to form the spiro-ether core.
- Finalize the synthesis via condensation with p-chlorobenzaldehyde under basic conditions to yield the final Lumefantrine Impurity 3 structure.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible benefits beyond mere technical feasibility. The primary advantage lies in the substantial cost savings achieved through the elimination of precious metal catalysts. Many modern coupling reactions rely on expensive palladium or rhodium complexes, which not only increase raw material costs but also necessitate complex downstream processing to remove toxic metal residues to ppm levels. In contrast, this patent utilizes commodity reagents like aluminum trichloride, sodium hydride, and sodium borohydride, which are inexpensive and globally available. This reliance on base metals and common reducing agents drastically simplifies the supply chain, reducing the risk of disruption caused by the volatility of the precious metals market and ensuring a more stable cost structure for long-term production contracts.
- Cost Reduction in Manufacturing: The economic viability of producing reference standards is often compromised by low yields and expensive reagents. This route mitigates those risks by using high-atom-economy reactions and avoiding proprietary catalysts. The avoidance of transition metals means that manufacturers do not need to invest in specialized scavenging resins or extensive filtration systems to meet residual metal specifications. Furthermore, the starting material, 2,7-dichlorofluorene, is a bulk chemical with a mature supply chain, preventing price gouging. These factors combine to significantly lower the Cost of Goods Sold (COGS), allowing suppliers to offer competitive pricing for high-purity reference standards without sacrificing margin.
- Enhanced Supply Chain Reliability: Supply continuity is critical for pharmaceutical quality control labs that cannot afford interruptions in their testing schedules. Because this synthesis relies on widely available organic solvents like ethanol, methanol, and ethyl acetate, there is minimal dependency on niche or regulated chemicals. The robustness of the reaction conditions—mostly operating within standard laboratory temperature ranges except for the specific lithiation step—means that the process can be transferred easily between different manufacturing sites. This flexibility ensures that even if one facility faces operational issues, production can be seamlessly shifted to another, guaranteeing reducing lead time for high-purity reference compounds and maintaining the integrity of the drug release schedule.
- Scalability and Environmental Compliance: As the demand for antimalarial drugs grows in endemic regions, the ability to scale impurity synthesis is vital. This five-step route is amenable to scale-up because it avoids hazardous reagents like diazomethane or highly unstable intermediates. The waste streams generated are primarily aqueous salts and organic solvents, which can be treated using standard effluent treatment protocols common in fine chemical plants. The process design inherently supports green chemistry principles by maximizing the utility of each atom in the starting materials and minimizing the generation of complex toxic byproducts. This alignment with environmental compliance standards simplifies the permitting process for commercial scale-up of complex organic intermediates, accelerating the time to market for the final reference standard.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Lumefantrine Impurity 3. These insights are derived directly from the patent specifications and are intended to assist R&D and quality assurance teams in evaluating the suitability of this material for their specific analytical needs. Understanding the synthesis and stability of this impurity is crucial for developing robust stability-indicating methods.
Q: Why is synthesizing Lumefantrine Impurity 3 critical for pharmaceutical manufacturers?
A: Regulatory bodies like the FDA and EMA require rigorous identification and quantification of impurities in antimalarial drugs like Lumefantrine to ensure patient safety. This patent provides a reliable method to generate the specific reference standard needed for HPLC calibration and quality control, filling a gap where no prior literature synthesis existed.
Q: What are the cost advantages of this new synthetic route compared to traditional isolation methods?
A: This route utilizes commercially available starting materials like 2,7-dichlorofluorene and avoids expensive transition metal catalysts. By relying on standard organic transformations such as Friedel-Crafts acylation and hydride reductions, the process significantly reduces raw material costs and eliminates the need for costly heavy metal scavenging steps often required in palladium-catalyzed couplings.
Q: Is this synthesis scalable for industrial production of reference standards?
A: Yes, the process employs robust reaction conditions and common solvents like dichloromethane, THF, and ethanol. While some steps require low-temperature control (e.g., -70°C for LDA addition), these are standard unit operations in fine chemical manufacturing, allowing for straightforward scale-up from gram-scale laboratory synthesis to multi-kilogram production runs.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Lumefantrine Impurity 3 Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your antimalarial drug products depends on the quality of your reference standards. Our team of expert chemists has extensively analyzed the synthetic pathway described in Patent CN116023387A and is fully prepared to execute this route with precision. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements whether for early-stage method development or full-scale QC validation. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of Lumefantrine Impurity 3 we deliver meets the exacting standards required by global pharmacopoeias.
We invite you to collaborate with us to secure your supply chain for this critical impurity. By leveraging our technical expertise, you can obtain a Customized Cost-Saving Analysis tailored to your specific consumption volumes. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. We are committed to supporting your regulatory success with high-quality intermediates that ensure the safety and efficacy of life-saving medications worldwide.