Scalable Synthesis of Fluorinated Pyrimidine Amines for Chronic Disease Drug Development
Scalable Synthesis of Fluorinated Pyrimidine Amines for Chronic Disease Drug Development
The pharmaceutical industry is constantly seeking robust and scalable pathways for synthesizing complex heterocyclic intermediates, particularly those containing fluorine motifs which are crucial for metabolic stability and bioavailability. Patent CN108395408B introduces a highly efficient preparation method for [4-(1,1-difluoro-ethyl)-pyrimidine]-5-methanamine hydrochloride, a valuable building block for new drug research targeting chronic conditions such as diabetes and alopecia. This technical disclosure outlines a streamlined three-step sequence starting from a pyrimidine aldehyde, utilizing an aqueous reduction followed by a Mitsunobu coupling and final deprotection. The strategic design of this route addresses common pain points in heterocyclic chemistry, such as solubility issues and functional group compatibility, offering a viable solution for reliable pharmaceutical intermediate supplier networks aiming to secure high-quality raw materials. By leveraging water as a reaction medium for the initial reduction step, the process significantly reduces the reliance on volatile organic compounds early in the synthesis, aligning with modern green chemistry principles while maintaining high reaction fidelity.
![Chemical structure of Compound M: [4-(1,1-difluoro-ethyl)-pyrimidine]-5-methanamine hydrochloride](/insights/img/difluoro-pyrimidine-amine-synthesis-pharma-supplier-20260305160640-01.png)
The structural integrity of the final product, as depicted in the associated chemical diagrams, highlights the precise placement of the difluoroethyl group at the 4-position and the aminomethyl group at the 5-position of the pyrimidine ring. This specific substitution pattern is often challenging to achieve via direct electrophilic substitution due to the electron-deficient nature of the pyrimidine core. The patented method circumvents these electronic limitations by constructing the side chain through nucleophilic transformations on a pre-functionalized scaffold. For R&D directors evaluating new routes for API manufacturing, this approach offers a distinct advantage in terms of impurity profile control, as the stepwise construction allows for rigorous purification of the alcohol and phthalimide intermediates before the final salt formation. The ability to produce this compound with high purity is essential for downstream biological screening, ensuring that observed pharmacological effects are attributable to the target molecule rather than synthetic byproducts.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes to fluorinated pyrimidine amines often rely on harsh halogenation followed by nucleophilic displacement with ammonia or amine equivalents, which can lead to poor regioselectivity and significant formation of poly-substituted byproducts. In many legacy processes, the introduction of the difluoroethyl moiety requires aggressive fluorinating agents like DAST at low temperatures, which poses significant safety hazards and scalability challenges in a commercial plant environment. Furthermore, conventional methods frequently utilize anhydrous organic solvents throughout the entire sequence, resulting in high solvent consumption and complex waste streams that increase the environmental footprint and disposal costs. The lack of chemoselectivity in older protocols often necessitates multiple chromatographic purification steps, which are impractical for multi-kilogram or ton-scale production, leading to substantial yield losses and extended cycle times. These inefficiencies create bottlenecks in the supply chain, making it difficult for procurement teams to secure consistent volumes of high-purity intermediates at a competitive price point.
The Novel Approach
The methodology described in CN108395408B represents a paradigm shift by employing a mild reduction strategy and a robust nitrogen installation technique that avoids the pitfalls of direct amination. By starting with 4-(1,1-difluoroethyl)-5-pyrimidine formaldehyde and reducing it with sodium borohydride in water, the process achieves excellent conversion rates under remarkably mild thermal conditions ranging from -10°C to 10°C. This aqueous workup not only simplifies the isolation of the alcohol intermediate but also minimizes the generation of hazardous waste associated with organic solvent extraction in the early stages. The subsequent use of the Mitsunobu reaction with phthalimide provides a controlled pathway to install the nitrogen atom with high specificity, effectively preventing over-alkylation or ring degradation that might occur with more aggressive aminating agents. This novel approach ensures cost reduction in API manufacturing by streamlining the purification workflow and utilizing reagents that are readily available on the global chemical market, thereby enhancing supply chain resilience.
Mechanistic Insights into Aqueous Borohydride Reduction and Mitsunobu Coupling
The core of this synthesis lies in the chemoselective reduction of the aldehyde functionality in the presence of the electron-withdrawing difluoroethyl group and the pyrimidine nitrogen atoms. When sodium borohydride is added portionwise to the aqueous suspension of the aldehyde at temperatures between -5°C and 5°C, the hydride ion selectively attacks the carbonyl carbon to form the corresponding alkoxide, which is then protonated by the solvent to yield 4-(1,1-difluoroethyl)-5-pyrimidine-methanol. The choice of water as a solvent is particularly ingenious; it suppresses potential side reactions such as Cannizzaro disproportionation which can occur in strongly basic organic media, and it facilitates the dissolution of inorganic borate byproducts, allowing for easy separation of the organic product. The stability of the difluoroethyl group under these reducing conditions is critical, as fluoride elimination is a common decomposition pathway for gem-difluoro compounds under basic conditions, yet the controlled pH and low temperature of this reaction preserve the integrity of the fluorine motif.
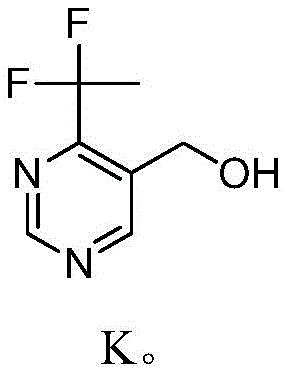
Following the formation of the alcohol intermediate, shown structurally as Compound K, the synthesis proceeds via a Mitsunobu reaction to convert the hydroxyl group into a protected amine. This transformation involves the activation of the alcohol by triphenylphosphine and diethyl azodicarboxylate (DEAD) to form an alkoxyphosphonium intermediate, which is then displaced by the phthalimide anion. The reaction is conducted in tetrahydrofuran (THF) under an inert atmosphere at temperatures not exceeding 30°C to prevent the decomposition of the azo reagent and to maintain the stereochemical integrity if chiral centers were present, although in this achiral system, it primarily ensures high yield and minimal byproduct formation. The use of phthalimide serves as a robust protecting group that withstands the reaction conditions and can be cleanly removed in the final step using hydrazine hydrate. This mechanistic pathway effectively isolates the nucleophilic nitrogen source from the electrophilic pyrimidine ring until the final deprotection, thereby minimizing the risk of N-alkylation on the ring nitrogens which would generate difficult-to-remove impurities.
How to Synthesize [4-(1,1-difluoro-ethyl)-pyrimidine]-5-methanamine Efficiently
To implement this synthesis effectively in a pilot or production setting, operators must adhere to strict temperature controls and reagent addition rates as defined in the patent examples. The process begins with the preparation of the aldehyde precursor, which can be derived from 4-ethyl-5-bromopyrimidine through a series of oxidation and fluorination steps, ultimately yielding the key starting material for the reduction. Once the aldehyde is secured, the reduction with sodium borohydride in water must be monitored closely via TLC to ensure complete consumption of the starting material before proceeding to extraction with dichloromethane. The detailed standardized synthesis steps for the subsequent Mitsunobu coupling and hydrazinolysis are critical for achieving the reported yields and purity levels, and these procedures have been optimized to balance reaction kinetics with operational safety.
- Perform reduction of 4-(1,1-difluoroethyl)-5-pyrimidine formaldehyde using sodium borohydride in water at -10°C to 10°C to obtain the alcohol intermediate.
- Execute a Mitsunobu reaction reacting the alcohol intermediate with triphenylphosphine, phthalimide, and diethyl azodicarboxylate in THF below 30°C.
- Conduct hydrazinolysis of the phthalimide derivative using hydrazine hydrate in ethanol under reflux to yield the final amine hydrochloride salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits in terms of operational efficiency and risk mitigation. The elimination of cryogenic conditions for the main transformation steps, relying instead on easily achievable cooling ranges like -10°C to 10°C, significantly lowers the energy consumption and equipment complexity required for production. This simplification translates directly into cost reduction in pharmaceutical intermediate manufacturing, as it allows for the utilization of standard glass-lined reactors without the need for specialized low-temperature infrastructure. Furthermore, the use of water as a primary solvent for the reduction step drastically reduces the volume of flammable organic solvents handled during the most exothermic part of the process, enhancing plant safety and reducing insurance and compliance costs associated with volatile organic compound (VOC) emissions.
- Cost Reduction in Manufacturing: The process avoids the use of expensive transition metal catalysts or exotic fluorinating reagents in the final amination steps, relying instead on commodity chemicals like sodium borohydride, triphenylphosphine, and hydrazine. This reliance on bulk chemicals ensures stable pricing and availability, shielding the supply chain from the volatility often seen with specialized catalytic systems. Additionally, the high selectivity of the Mitsunobu reaction minimizes the formation of isomeric impurities, reducing the need for costly preparative HPLC or repeated recrystallizations, which are major cost drivers in fine chemical production. The overall atom economy and yield optimization inherent in this route contribute to a lower cost of goods sold (COGS), enabling more competitive pricing for the final API.
- Enhanced Supply Chain Reliability: By utilizing a linear and robust synthetic sequence, the risk of batch failure due to sensitive reaction conditions is markedly decreased. The intermediates, such as the phthalimide derivative, are stable solids that can be isolated, characterized, and stored, providing flexibility in production scheduling and inventory management. This modularity allows manufacturers to stockpile key intermediates during periods of low demand or raw material abundance, ensuring continuous supply even if upstream feedstock availability fluctuates. The straightforward workup procedures, involving standard extractions and filtrations, facilitate rapid turnover of reactor vessels, increasing the overall throughput capacity of the manufacturing facility and reducing lead times for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The process is designed with industrial scale-up in mind, evidenced by the use of solvents like ethanol and THF which are easily recovered and recycled via distillation. The aqueous nature of the first step simplifies effluent treatment, as the primary waste stream contains inorganic borates which are less hazardous than heavy metal residues or chlorinated organic wastes. This alignment with environmental regulations reduces the burden on waste treatment facilities and minimizes the risk of regulatory shutdowns, ensuring long-term operational continuity. The ability to scale from gram to kilogram quantities without fundamental changes to the reaction parameters demonstrates the commercial viability of this route for meeting the growing demand for fluorinated pyrimidine therapeutics.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this patented synthesis route. Understanding these nuances is vital for process chemists and engineers tasked with transferring this technology from the laboratory to the production floor. The answers provided are derived directly from the experimental data and specifications outlined in the patent documentation, ensuring accuracy and relevance for technical decision-making.
Q: What are the critical temperature controls for the sodium borohydride reduction step?
A: The patent specifies a strict temperature range of -10°C to 10°C, preferably -5°C to 5°C, during the addition of sodium borohydride to ensure selective reduction of the aldehyde without affecting the difluoroethyl group or the pyrimidine ring stability.
Q: Why is the Mitsunobu reaction preferred for introducing the amine functionality in this route?
A: The Mitsunobu protocol using phthalimide allows for the conversion of the primary alcohol to a protected amine under mild conditions (below 30°C), avoiding harsh acidic or basic conditions that could degrade the sensitive fluorinated pyrimidine scaffold.
Q: How does this synthesis route support industrial scale-up compared to traditional methods?
A: The process utilizes water as a solvent for the initial reduction and common organic solvents like THF and ethanol for subsequent steps, eliminating the need for cryogenic conditions or exotic catalysts, thereby simplifying waste treatment and enhancing operational safety for large-scale production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable [4-(1,1-difluoro-ethyl)-pyrimidine]-5-methanamine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of next-generation therapeutics for chronic diseases. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from bench-scale discovery to full-scale manufacturing is seamless and efficient. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs that employ advanced analytical techniques to verify the identity and quality of every batch. Our capability to handle complex fluorinated heterocycles positions us as a strategic partner for pharmaceutical companies seeking to accelerate their drug development timelines without compromising on quality or compliance.
We invite you to collaborate with us to leverage this innovative synthetic route for your specific project needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating how our optimized processes can enhance your bottom line. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise in fine chemical synthesis can support your supply chain goals and drive your research forward.