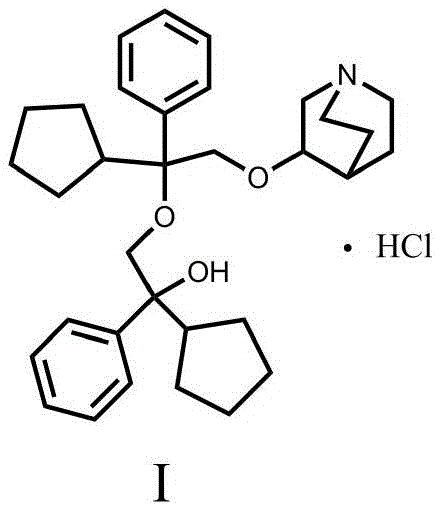
Advanced Synthesis of Penehyclidine Hydrochloride Impurity for Quality Control
Advanced Synthesis of Penehyclidine Hydrochloride Impurity for Quality Control
The rigorous demand for pharmaceutical quality control has necessitated the development of robust synthetic routes for critical impurities, particularly for potent anticholinergic agents like penehyclidine hydrochloride. Patent CN109824661B introduces a groundbreaking preparation method for a specific dimeric impurity, chemically defined as 3-[2-cyclopentyl-2-phenyl-2-(2-cyclopentyl-2-hydroxy-2-phenyl-ethoxy)ethoxy]quinuclidine hydrochloride. This compound represents a major process-related impurity that must be strictly monitored to ensure patient safety and regulatory compliance. The disclosed technology leverages a novel epoxide ring-opening strategy that fundamentally alters the efficiency profile of impurity synthesis, moving away from laborious purification techniques toward a streamlined, high-yield process. By utilizing alpha-phenyl-alpha-cyclopentyl-ethylene oxide as a key building block, the method achieves exceptional purity profiles while maintaining operational simplicity. For R&D teams and quality assurance managers, access to such high-fidelity reference standards is non-negotiable for validating analytical methods and establishing stability-indicating assays.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of this specific impurity relied heavily on methodologies described in earlier patents such as CN106518862A, which utilized alpha-phenyl-alpha-cyclopentyl-alpha-hydroxy-p-toluenesulfonic acid ethyl ester as the electrophilic partner. This traditional approach suffers from inherent kinetic and thermodynamic limitations that plague large-scale operations. The reaction often proceeds incompletely due to steric hindrance associated with the bulky tosylate leaving group, leading to a complex mixture of unreacted starting materials and side products. Furthermore, the isolation of the desired product typically necessitates column chromatography, a technique that is notoriously difficult to scale, expensive in terms of silica gel consumption, and time-consuming regarding solvent recovery. The cumulative effect of these inefficiencies results in significantly depressed yields and extended production cycles, creating a bottleneck for laboratories requiring substantial quantities of the impurity for toxicological studies or method validation. The reliance on such cumbersome purification protocols also introduces variability in batch-to-batch consistency, which is detrimental when preparing certified reference materials.
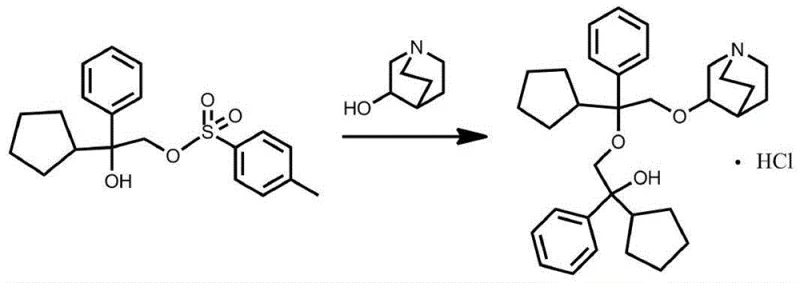
The Novel Approach
In stark contrast, the methodology outlined in CN109824661B employs a direct nucleophilic substitution using an epoxide derivative, which offers a superior leaving group capability and reduced steric congestion during the transition state. This strategic shift allows the reaction to proceed with high conversion rates under relatively mild alkaline conditions, effectively bypassing the formation of stubborn byproducts that characterize the tosylate route. The process flow is drastically simplified; instead of chromatographic separation, the product can be isolated through standard liquid-liquid extraction followed by crystallization, operations that are inherently scalable and cost-effective. The use of dimethyl sulfoxide (DMSO) as the reaction medium further enhances the solubility of the polar intermediates, ensuring homogeneous reaction conditions that promote uniform product formation. This novel approach not only accelerates the timeline from raw material to finished standard but also aligns perfectly with green chemistry principles by minimizing solvent waste and eliminating the need for disposable silica media. Consequently, this method stands as a reliable solution for the cost reduction in pharmaceutical intermediate manufacturing, specifically for high-value impurity standards.
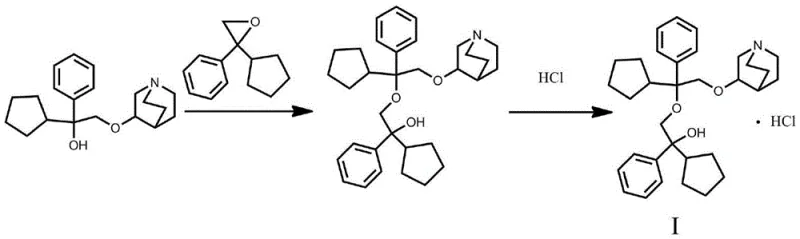
Mechanistic Insights into Base-Catalyzed Epoxide Ring Opening
The core of this synthetic breakthrough lies in the base-catalyzed nucleophilic attack of the quinuclidine hydroxyl group on the strained three-membered epoxide ring. Under the influence of strong bases such as sodium hydride (NaH), the hydroxyl proton of penehyclidine is abstracted to form a highly reactive alkoxide species. This alkoxide acts as a potent nucleophile, seeking out the electrophilic carbons of the alpha-phenyl-alpha-cyclopentyl-ethylene oxide. The ring strain of the epoxide provides the necessary driving force for the reaction, allowing the bond to break and the new ether linkage to form with high regioselectivity. The presence of the phenyl and cyclopentyl groups on the epoxide influences the electronic distribution, yet the reaction conditions (60-80°C) provide sufficient thermal energy to overcome activation barriers without inducing significant degradation. This mechanistic pathway is far more direct than the SN2 displacement required in the tosylate method, as it avoids the generation of sulfonate salts that can complicate downstream processing. Understanding this mechanism is crucial for process chemists aiming to optimize reaction parameters for commercial scale-up of complex pharmaceutical intermediates.
Impurity control is intrinsically linked to the selectivity of this ring-opening event. The optimized molar ratios, specifically maintaining a slight excess of the epoxide (1.05 to 1.1 equivalents) relative to the base and penehyclidine, ensure that the nucleophile is fully consumed, thereby minimizing the presence of unreacted starting material in the final matrix. Additionally, the choice of sodium hydride over weaker bases ensures rapid and complete deprotonation, reducing the window of time available for competing side reactions such as epoxide polymerization or hydrolysis. The subsequent acidification step with hydrogen chloride gas is carefully controlled to a pH of 1-2, which facilitates the precipitation of the hydrochloride salt while keeping soluble organic impurities in the mother liquor. This precise control over the salification process is what enables the achievement of purity levels exceeding 99%, a critical benchmark for any substance intended for use as a quantitative standard in HPLC or GC analysis. Such rigorous control mechanisms demonstrate why this patent represents a significant advancement in high-purity OLED material and pharmaceutical intermediate synthesis standards.
How to Synthesize 3-[2-cyclopentyl-2-phenyl-2-(2-cyclopentyl-2-hydroxy-2-phenyl-ethoxy)ethoxy]quinuclidine Efficiently
The practical execution of this synthesis requires careful attention to moisture control and temperature management to maximize the efficacy of the sodium hydride reagent. The process begins with the dissolution of penehyclidine in anhydrous DMSO, followed by the cautious addition of the base to generate the alkoxide in situ. Once the reactive species is formed, the epoxide is introduced dropwise to manage the exotherm and maintain selectivity. Following the reaction period, a straightforward aqueous workup allows for the separation of the organic product, which is then converted to its stable hydrochloride salt form through gas sparging and crystallization. This sequence eliminates the need for specialized equipment beyond standard reactor vessels, making it accessible for both laboratory and pilot plant environments. For detailed operational parameters and safety guidelines, the standardized synthesis steps are provided below.
- Dissolve penehyclidine in DMSO, add sodium hydride, and react with alpha-phenyl-alpha-cyclopentyl-ethylene oxide at 60-80°C.
- Extract the resulting free base with methyl tert-butyl ether and concentrate to obtain the viscous oil intermediate.
- Dissolve the free base in an organic solvent, introduce hydrogen chloride gas to adjust pH to 1-2, and crystallize to obtain the final hydrochloride salt.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the adoption of this epoxide-based synthesis route offers transformative benefits that directly impact the bottom line and supply chain resilience. The elimination of column chromatography is perhaps the most significant economic driver, as it removes a major cost center associated with silica gel procurement, solvent consumption, and labor-intensive fraction collection. This simplification translates into substantially reduced manufacturing costs, allowing for more competitive pricing of the final impurity standard without compromising on quality. Furthermore, the raw materials required, such as the epoxide derivative and common inorganic bases, are commodity chemicals that are readily available from multiple global suppliers, mitigating the risk of single-source dependency. This availability ensures that production schedules can be maintained consistently, reducing lead time for high-purity pharmaceutical intermediates even during periods of market volatility. The robustness of the process also means that batch failure rates are minimized, providing supply chain heads with greater predictability in inventory planning and delivery commitments.
- Cost Reduction in Manufacturing: The removal of chromatographic purification steps drastically lowers the operational expenditure per kilogram of product produced. By relying on crystallization and extraction, the process utilizes cheaper solvents and reduces waste disposal costs associated with silica gel. This efficiency gain allows for significant cost savings that can be passed down to the customer or reinvested into further R&D initiatives. Additionally, the higher yield obtained through this method means that less raw material is wasted, further enhancing the overall economic viability of the production campaign.
- Enhanced Supply Chain Reliability: The use of widely available starting materials ensures that production is not held hostage by the scarcity of exotic reagents. The simplified workflow reduces the complexity of the manufacturing schedule, allowing for faster turnaround times from order to shipment. This agility is crucial for pharmaceutical companies that require urgent delivery of reference standards to meet regulatory filing deadlines. The stability of the process parameters also ensures that the supply remains continuous and consistent, fostering long-term partnerships between suppliers and manufacturers.
- Scalability and Environmental Compliance: The transition from batch chromatography to crystallization makes this process inherently scalable from gram to ton quantities without fundamental changes to the unit operations. This scalability supports the growing demand for impurity standards as drug volumes increase in the market. Moreover, the reduction in solvent usage and solid waste generation aligns with increasingly stringent environmental regulations, reducing the carbon footprint of the manufacturing process. This environmental compliance is a key factor for multinational corporations seeking sustainable supply chain partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of this penehyclidine impurity. These answers are derived directly from the experimental data and beneficial effects reported in the patent literature, ensuring accuracy and relevance for technical decision-makers. Understanding these details helps stakeholders evaluate the suitability of this material for their specific quality control workflows. For further customization or bulk inquiries, our technical team is prepared to provide deeper insights.
Q: What is the primary advantage of using the epoxide route over the tosylate route?
A: The epoxide route eliminates the need for complex column chromatography purification, significantly simplifying post-treatment and improving overall yield compared to the traditional tosylate method.
Q: What purity levels can be achieved with this synthesis method?
A: The described method consistently achieves purity levels exceeding 99%, making it highly suitable for generating reference standards for quality control.
Q: Is this process scalable for industrial production?
A: Yes, the process utilizes cheap and easily obtained raw materials and avoids difficult separation steps, making it well-suited for mass preparation and commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-[2-cyclopentyl-2-phenyl-2-(2-cyclopentyl-2-hydroxy-2-phenyl-ethoxy)ethoxy]quinuclidine hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your analytical data depends entirely on the quality of your reference standards. Our expertise in organic synthesis allows us to replicate and optimize complex pathways like the one described in CN109824661B, ensuring that we deliver materials that meet the highest industry benchmarks. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, guaranteeing that we can meet your volume requirements whether for early-stage research or full-scale QC operations. Our facilities are equipped with rigorous QC labs and advanced analytical instrumentation to verify stringent purity specifications, ensuring that every batch of impurity standard we supply is accompanied by comprehensive characterization data. This commitment to quality makes us a trusted partner for pharmaceutical companies worldwide who cannot afford ambiguity in their impurity profiling.
We invite you to leverage our technical capabilities to streamline your supply chain and reduce your overall cost of goods. By choosing NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume needs and purity requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments for this and other critical pharmaceutical intermediates. Let us help you secure a stable, high-quality supply of essential reference materials that support your mission to bring safe and effective medicines to market.