Streamlined Manufacturing of High-Purity Anti-HCV Phosphoramidate Intermediates for Global Supply
Streamlined Manufacturing of High-Purity Anti-HCV Phosphoramidate Intermediates for Global Supply
The global demand for direct-acting antiviral agents, particularly those targeting Hepatitis C Virus (HCV), continues to drive innovation in pharmaceutical intermediate manufacturing. A pivotal advancement in this sector is detailed in patent CN112940053B, which discloses a highly efficient preparation method for anti-HCV medicaments, specifically focusing on nucleoside phosphoramidate prodrugs similar to the Sofosbuvir class. This technology addresses critical bottlenecks in the supply chain by offering a synthetic route that is not only concise but also operationally robust, ensuring consistent quality for downstream API production. For R&D directors and procurement strategists, understanding the nuances of this pathway is essential for securing a reliable pharmaceutical intermediate supplier capable of meeting rigorous purity standards while optimizing cost structures.
The significance of this patent lies in its ability to bypass traditional synthetic hurdles that have historically plagued the production of complex nucleoside analogs. By leveraging specific Lewis acid catalysis and optimized coupling conditions, the method achieves exceptional yields and purity profiles that are directly translatable to commercial scale. This report provides a deep technical analysis of the methodology, contrasting it with legacy processes to highlight the tangible benefits for cost reduction in API manufacturing and supply chain resilience.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of nucleoside phosphoramidates has been fraught with inefficiencies, primarily due to the necessity of extensive protecting group strategies. As illustrated in prior art references such as WO2008045419 and WO2011123668, traditional routes often involve the installation of benzoyl or other protecting groups on the sugar moiety to prevent side reactions during phosphorylation and glycosylation steps.
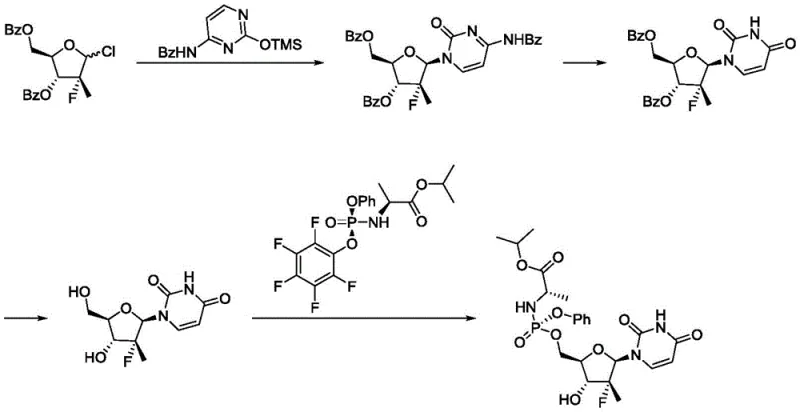
These conventional pathways typically require a sequence where the sugar hydroxyls are protected, the nucleobase is coupled, the phosphate moiety is installed, and finally, a global deprotection is performed. This multi-step sequence not only extends the overall timeline but also introduces significant yield losses at each transformation. Furthermore, the reliance on column chromatography for purification in these older methods creates a major bottleneck for commercial scale-up of complex pharmaceutical intermediates, as chromatographic separation is notoriously difficult to implement efficiently at the multi-kilogram or metric-ton level. The cumulative effect is a process with high operational expenditure (OPEX) and a larger environmental footprint due to excessive solvent usage.
The Novel Approach
In stark contrast, the methodology disclosed in patent CN112940053B presents a streamlined, two-step strategy that fundamentally rethinks the order of operations. The core innovation involves the direct phosphorylation of a fluorinated sugar halide followed by the coupling of the nucleobase, effectively eliminating the need for temporary hydroxyl protection groups.
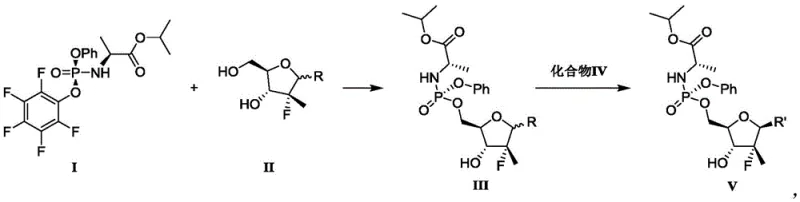
This novel approach utilizes a specific phosphoramidate reagent (Compound I) which reacts directly with a sugar halide (Compound II) bearing free hydroxyl groups. The reaction is mediated by a Lewis acid and a base, selectively installing the phosphoramidate moiety at the 5'-position while leaving the anomeric halide intact for subsequent coupling. This strategic inversion of the synthetic sequence allows for the construction of the complex phosphoramidate architecture with minimal handling. The result is a process that is not only shorter but also inherently safer and more scalable, positioning it as a superior choice for any reliable pharmaceutical intermediate supplier aiming to optimize their portfolio.
Mechanistic Insights into Lewis Acid-Catalyzed Phosphorylation and Coupling
The success of this synthetic route hinges on the precise control of chemoselectivity during the phosphorylation step. The reaction between Compound I and Compound II is facilitated by Lewis acids such as zinc chloride, anhydrous magnesium chloride, or tin tetrachloride. These metal centers coordinate with the oxygen atoms of the phosphoramidate and the sugar hydroxyls, activating the phosphorus center for nucleophilic attack by the 5'-hydroxyl group of the sugar. The presence of a base, such as triethylamine or diisopropylethylamine, serves to neutralize the acid byproduct generated during the substitution, driving the equilibrium forward. This mechanism ensures high regioselectivity, preventing unwanted phosphorylation at the 3'-hydroxyl position, which is critical for maintaining the biological activity of the final drug substance.
Following the formation of the phosphorylated intermediate (Compound III), the second critical transformation is the glycosylation or coupling with the nucleobase (Compound IV). This step employs conditions reminiscent of the Vorbrüggen coupling but adapted for the sensitive phosphoramidate substrate. As shown in specific embodiments, the nucleobase is often silylated in situ using reagents like N,O-bis(trimethylsilyl)acetamide (BSA) to enhance its nucleophilicity.
The coupling is then promoted by a strong Lewis acid, typically tin tetrachloride, at elevated temperatures (reflux in chlorobenzene or dichloromethane). This activates the anomeric halide, facilitating the displacement by the silylated nucleobase to form the beta-anomer with high stereoselectivity. The robustness of this mechanism is evidenced by the ability to tolerate various halides (Cl, Br, I) at the anomeric center and different nucleobases, including uracil derivatives and purine analogs, yielding products with purity exceeding 99% after simple crystallization. This level of impurity control is paramount for meeting the stringent specifications required by regulatory bodies for antiviral therapies.
How to Synthesize Anti-HCV Phosphoramidate Intermediates Efficiently
Implementing this synthesis requires careful attention to reaction parameters, particularly the stoichiometry of the Lewis acid and the choice of solvent to ensure optimal solubility and reaction kinetics. The process is designed to be telescoped where possible, minimizing isolation steps and maximizing throughput. For detailed operational protocols, including specific molar ratios, temperature profiles, and workup procedures derived from the patent examples, please refer to the standardized guide below.
- React a phosphoramidate reagent (Compound I) with a fluorinated sugar halide (Compound II) in the presence of a Lewis acid (e.g., ZnCl2, MgCl2) and a base to form the phosphorylated intermediate (Compound III).
- Couple the phosphorylated intermediate (Compound III) with a nucleobase (Compound IV, such as a uracil or purine derivative) using a condensing agent like tin tetrachloride to yield the final anti-HCV drug intermediate (Compound V).
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers profound advantages that extend beyond mere technical elegance. For procurement managers and supply chain heads, the primary value proposition lies in the drastic simplification of the manufacturing workflow, which directly correlates to reduced lead times and lower total landed costs. By eliminating the protection and deprotection cycles, the number of unit operations is significantly reduced, thereby decreasing the consumption of raw materials, solvents, and energy. This efficiency gain translates into substantial cost savings that can be passed down the supply chain, making the final API more competitive in the global market.
- Cost Reduction in Manufacturing: The elimination of benzoyl protection and subsequent deprotection steps removes the need for expensive reagents like benzoyl chloride and the associated waste disposal costs. Furthermore, the shift from column chromatography to crystallization for purification represents a massive reduction in processing time and silica gel consumption. Crystallization is a standard unit operation in chemical engineering that scales linearly and predictably, unlike chromatography which often requires complex simulated moving bed (SMB) systems for large volumes. This transition ensures that the cost reduction in pharmaceutical intermediate manufacturing is structural and sustainable, rather than dependent on volatile raw material pricing.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions—operating at mild temperatures such as 25°C for phosphorylation and moderate reflux for coupling—reduces the risk of batch failures due to thermal runaway or sensitivity. The use of common, commercially available solvents like tetrahydrofuran, ethyl acetate, and dichloromethane ensures that supply disruptions are unlikely. Additionally, the high yields reported (consistently above 90% in key steps) mean that less starting material is required to produce the same amount of product, buffering the supply chain against upstream shortages of specialized sugar precursors.
- Scalability and Environmental Compliance: The process generates significantly less hazardous waste compared to traditional routes. By avoiding heavy metal catalysts in favor of zinc or magnesium salts, and reducing solvent intensity through higher concentrations and crystallization, the environmental footprint is minimized. This aligns with increasingly strict global environmental regulations, reducing the compliance burden on manufacturing sites. The ability to scale from laboratory grams to multi-ton production without re-engineering the core chemistry provides a clear pathway for reducing lead time for high-purity pharmaceutical intermediates, ensuring continuous availability for downstream formulation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity for technical teams evaluating this route for adoption.
Q: How does this new synthesis route improve upon conventional methods for anti-HCV intermediates?
A: Conventional routes often require multiple protection and deprotection steps (e.g., benzoyl groups) and column chromatography purification, which increases cost and complexity. This novel method utilizes a direct phosphorylation strategy on a sugar halide followed by coupling, eliminating protection steps and enabling purification via simple crystallization, significantly enhancing industrial feasibility.
Q: What specific catalysts are employed in this phosphoramidate synthesis?
A: The process utilizes mild Lewis acids such as zinc chloride, anhydrous magnesium chloride, or tin tetrachloride in combination with organic bases like triethylamine or DIPEA. These conditions allow for high yields (up to 93%) and excellent purity (over 99%) without harsh reaction environments.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process is specifically designed for industrial scale-up. It operates at mild temperatures (25°C to 80°C), uses common solvents like THF and dichloromethane, and avoids difficult purification techniques like column chromatography, relying instead on crystallization which is ideal for metric-ton manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Anti-HCV Intermediate Supplier
The technological breakthroughs encapsulated in patent CN112940053B represent a significant leap forward in the synthesis of antiviral nucleotides. However, translating patent claims into consistent, commercial-grade supply requires a partner with deep process engineering expertise. NINGBO INNO PHARMCHEM stands at the forefront of this capability, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facilities are equipped with state-of-the-art reactors and rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of phosphoramidate intermediate meets the exacting standards required for GMP API synthesis.
We invite global pharmaceutical partners to leverage our technical proficiency to secure their supply chains. By collaborating with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, ensuring that your next-generation HCV therapies reach the market faster and more efficiently.