Advanced Synthesis of 2,6-Diazaspiro[3.5]nonane Intermediates for Commercial Scale-Up
Advanced Synthesis of 2,6-Diazaspiro[3.5]nonane Intermediates for Commercial Scale-Up
The pharmaceutical industry continuously demands robust and scalable synthetic routes for complex heterocyclic scaffolds, particularly spirocyclic amines which are prevalent in modern kinase inhibitors and CNS drugs. Patent CN113214255A introduces a highly efficient methodology for the synthesis of 2,6-diazaspiro[3.5]nonane-6-tert-butyl formate and its salts, addressing critical bottlenecks in existing literature. This technical disclosure outlines a streamlined four-step sequence that transforms readily available precursors into high-value intermediates with exceptional operational simplicity. By leveraging a strategic combination of nucleophilic substitution, reductive cyclization, and protective group chemistry, this process offers a viable pathway for reliable pharmaceutical intermediate supplier networks seeking to optimize their supply chains. The following analysis dissects the chemical ingenuity and commercial potential embedded within this patent, providing R&D and procurement leaders with actionable insights into adopting this technology for commercial scale-up of complex pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of 2,6-diazaspiro[3.5]nonane cores has been plagued by multi-step sequences that suffer from poor atom economy and the requirement for exotic or prohibitively expensive starting materials. Traditional approaches often rely on complex cyclization strategies that demand harsh reaction conditions, leading to significant impurity profiles that are difficult to purge during downstream processing. Furthermore, many legacy routes utilize protecting group strategies that are not orthogonal, necessitating additional synthetic steps to install and remove masking groups, which drastically reduces the overall yield and increases the cost of goods sold. The reliance on transition metals that are difficult to remove to ppm levels also poses a significant regulatory hurdle for API manufacturers, complicating the validation process and extending lead times for high-purity pharmaceutical intermediates. These inefficiencies create a fragile supply chain where minor deviations in raw material quality can result in batch failures, undermining the reliability required for GMP manufacturing environments.
The Novel Approach
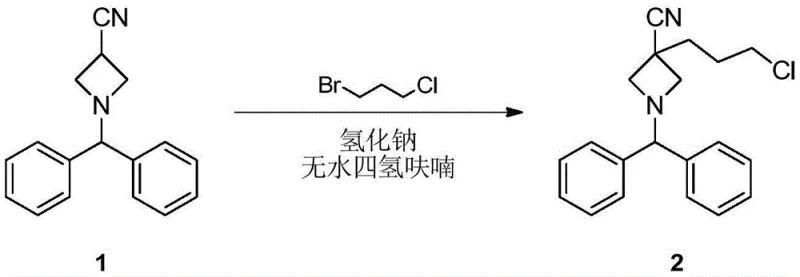
In stark contrast, the methodology described in CN113214255A presents a paradigm shift by utilizing 1-benzhydryl-3-cyanoazetidine as a foundational building block, a material that is both commercially accessible and cost-effective. The innovation lies in the telescoping of the ring-closing event with the reduction of the nitrile functionality, effectively merging two critical transformations into a single operational step. This design eliminates the need for isolating unstable amino-intermediates, thereby reducing exposure to air and moisture which can degrade product quality. The route is characterized by mild reaction conditions, predominantly operating between -80°C and 50°C, which enhances safety profiles and allows for the use of standard stainless steel reactors without specialized lining. By shortening the synthetic linear sequence to just four steps while maintaining high selectivity, this approach significantly mitigates the risk of yield erosion, ensuring that the final output meets the stringent purity specifications demanded by top-tier drug developers.
Mechanistic Insights into Reductive Cyclization and Spiro-Formation
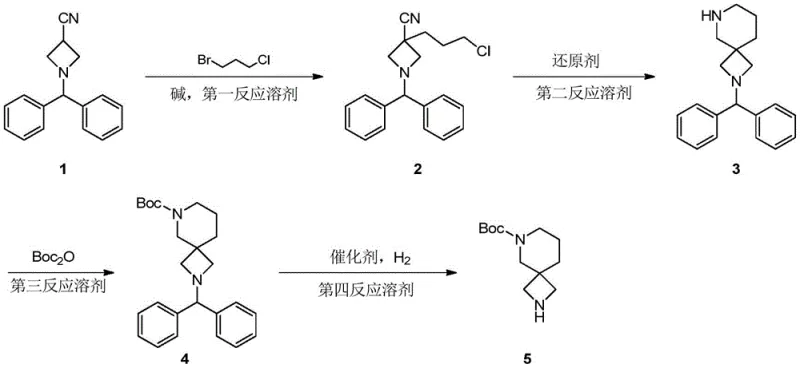
The cornerstone of this synthetic strategy is the second step, where the linear precursor undergoes a transformative reductive cyclization to establish the spirocyclic architecture. As illustrated in the reaction scheme below, the process initiates with the reduction of the cyano group on the azetidine ring using lithium aluminum hydride (LiAlH4) in anhydrous tetrahydrofuran. This powerful reducing agent converts the nitrile moiety into a primary amine in situ. Crucially, the molecule is pre-functionalized with a terminal chloro-propyl chain attached to the azetidine nitrogen from the previous alkylation step. Once the primary amine is generated, it acts as an internal nucleophile, attacking the terminal carbon bearing the chlorine atom. This intramolecular SN2 displacement occurs spontaneously under the reaction conditions, closing the six-membered piperidine ring and forming the stable 2,6-diazaspiro[3.5]nonane core. This tandem reduction-cyclization mechanism is highly efficient as it avoids the isolation of reactive diamine species, minimizing side reactions such as polymerization or intermolecular coupling.
Following the formation of the spiro-cycle, the synthesis proceeds with the installation of the tert-butoxycarbonyl (Boc) protecting group on the newly formed piperidine nitrogen. This step is critical for differentiating the reactivity of the two nitrogen atoms within the spiro system for subsequent functionalization in drug discovery campaigns. The use of di-tert-butyl dicarbonate (Boc2O) in alcoholic solvents like methanol or ethanol ensures high conversion rates under ambient temperatures, preserving the integrity of the sensitive spiro-scaffold. The final stage involves the removal of the benzhydryl protecting group via catalytic hydrogenation. Utilizing palladium on carbon (Pd/C) under hydrogen pressure in the presence of acetic acid facilitates the cleavage of the C-N bond, unmasking the secondary amine on the azetidine ring. This hydrogenolysis step is remarkably clean, producing toluene as a benign byproduct that is easily removed during workup, thus contributing to a superior impurity profile and facilitating easier purification protocols for the final API intermediate.
How to Synthesize 2,6-Diazaspiro[3.5]nonane Derivatives Efficiently
Executing this synthesis requires precise control over stoichiometry and temperature to maximize the efficiency of the alkylation and cyclization steps. The process begins with the generation of a lithiated species using LDA or sodium hydride, followed by the addition of the alkylating agent. Subsequent reduction with LiAlH4 must be managed carefully to control exotherms, followed by a standard Boc protection and final hydrogenolysis. For laboratory and pilot plant teams aiming to replicate these results, adherence to the specific molar ratios and solvent choices outlined in the patent is essential to achieve the reported yields.
- Perform nucleophilic substitution on 1-benzhydryl-3-cyanoazetidine with 1-bromo-3-chloropropane using LDA base in anhydrous THF at low temperatures.
- Reduce the cyano group and induce intramolecular cyclization using lithium aluminum hydride in tetrahydrofuran to form the spiro-piperidine core.
- Protect the secondary amine with di-tert-butyl dicarbonate (Boc2O) in methanol or ethanol, followed by catalytic hydrogenation to remove the benzhydryl group.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this synthetic route offers substantial cost reduction in pharmaceutical intermediate manufacturing by relying on commodity chemicals rather than custom-synthesized building blocks. The primary starting materials, including 1-bromo-3-chloropropane and benzhydryl-protected azetidines, are available in bulk quantities from multiple global suppliers, mitigating the risk of single-source dependency. This diversification of the supply base enhances supply chain reliability, ensuring that production schedules are not disrupted by raw material shortages. Furthermore, the elimination of complex chromatographic purifications in favor of crystallization and extraction techniques significantly lowers the operational expenditure associated with solvent consumption and waste disposal. The robustness of the reaction conditions means that the process is less sensitive to minor fluctuations in utility parameters, such as cooling water temperature, which translates to higher batch success rates and consistent throughput in a commercial setting.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the high atom economy of the cyclization step and the use of inexpensive reagents like LiAlH4 and Boc2O. By combining the reduction and ring-closing into a single pot, the method eliminates an entire unit operation, reducing labor costs, energy consumption, and equipment occupancy time. Additionally, the avoidance of precious metal catalysts in the early stages, reserving palladium only for the final deprotection where it can be efficiently recovered, further optimizes the cost structure. These factors collectively contribute to a lower cost of goods, allowing procurement managers to negotiate more competitive pricing for long-term supply agreements without compromising on quality standards.
- Enhanced Supply Chain Reliability: The simplicity of the four-step sequence inherently reduces the lead time for high-purity pharmaceutical intermediates by minimizing the number of quality control checkpoints and intermediate storage requirements. Since the route does not depend on cryogenic conditions below -80°C for extended periods (only for initial deprotonation), it can be executed in standard multipurpose plants without requiring specialized low-temperature infrastructure. This flexibility allows manufacturers to allocate production slots more efficiently, responding rapidly to fluctuating market demands. The stability of the Boc-protected intermediate also permits strategic stockpiling, providing a buffer against supply chain volatility and ensuring continuity of supply for downstream API synthesis.
- Scalability and Environmental Compliance: Scaling this process from kilogram to metric ton quantities is facilitated by the use of common organic solvents like THF, ethanol, and ethyl acetate, which have well-established recovery and recycling protocols. The waste streams generated are primarily aqueous salts and organic solvents that can be treated using conventional effluent treatment plants, ensuring compliance with increasingly stringent environmental regulations. The high selectivity of the reactions minimizes the formation of hazardous byproducts, reducing the burden on EHS teams. Moreover, the final hydrogenation step operates at moderate pressures (20-100 psi), which is well within the safety limits of standard industrial hydrogenation reactors, removing barriers to rapid capacity expansion.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this synthesis method, derived from the specific embodiments and claims within the patent documentation. Understanding these nuances is vital for process chemists evaluating the feasibility of technology transfer.
Q: What are the key advantages of this synthesis route compared to traditional methods?
A: This method utilizes commercially available and inexpensive raw materials like 1-benzhydryl-3-cyanoazetidine. It features a short four-step route with a clever one-pot reduction-cyclization step, significantly simplifying the process and improving overall yield suitability for industrial production.
Q: How is the spirocyclic ring formed in this process?
A: The spirocyclic structure is formed during the second step via an intramolecular cyclization. After the nitrile group is reduced to a primary amine by lithium aluminum hydride, the newly formed amine nucleophilically attacks the terminal chloride of the propyl chain, closing the piperidine ring spontaneously.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the patent explicitly states the method is suitable for amplification and industrial production. The reaction conditions are safe and easy to control, utilizing standard solvents like THF and ethanol, and common catalysts like palladium on carbon, which facilitates scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2,6-Diazaspiro[3.5]nonane Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality spirocyclic intermediates play in the development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering products that meet stringent purity specifications through our rigorous QC labs, which employ state-of-the-art analytical instrumentation to verify identity and assay. By partnering with us, clients gain access to a supply chain that is not only robust and compliant but also optimized for speed and cost-efficiency, aligning perfectly with the aggressive timelines of modern drug development programs.
We invite you to engage with our technical procurement team to discuss how this patented synthesis route can be tailored to your specific project needs. Whether you require a Customized Cost-Saving Analysis for your current supply chain or need to obtain specific COA data and route feasibility assessments for this spirocyclic scaffold, we are ready to provide the support necessary to accelerate your pipeline. Contact us today to explore how our manufacturing capabilities can drive value and reliability for your organization.