Scalable Manufacturing of Alkynyl Pyridine Prolyl Hydroxylase Inhibitors via Optimized Sonogashira Coupling
Scalable Manufacturing of Alkynyl Pyridine Prolyl Hydroxylase Inhibitors via Optimized Sonogashira Coupling
The pharmaceutical industry is constantly seeking robust manufacturing pathways for complex small molecules, particularly those targeting hypoxia-inducible factor (HIF) stabilization. Patent CN111094246B discloses a significant advancement in the preparation of alkynyl pyridine prolyl hydroxylase (PHD) inhibitors, which are critical therapeutic candidates for treating anemia and ischemic diseases such as chronic kidney disease. This technical insight report analyzes the transition from laboratory-scale curiosity to industrial viability, focusing on the strategic replacement of hazardous reagents and non-scalable unit operations. By leveraging a protected intermediate strategy combined with optimized Sonogashira coupling conditions, this methodology offers a reliable pharmaceutical intermediates supplier pathway that addresses both purity and throughput concerns. The following analysis details how this specific intellectual property provides a foundation for cost reduction in pharmaceutical intermediates manufacturing while maintaining stringent quality control standards required by global regulatory bodies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
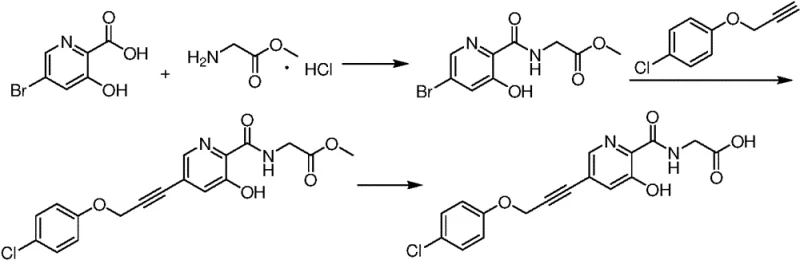
Prior art methods, such as those disclosed in CN105130888A, relied heavily on reaction conditions that are fundamentally incompatible with large-scale commercial production. A primary bottleneck was the utilization of microwave irradiation for the key coupling step, a technology that poses severe challenges for heat transfer and penetration depth when scaling beyond gram quantities. Furthermore, the conventional route employed 1-hydroxybenzotriazole (HOBT) as a condensing agent, a reagent known for its potential explosiveness in anhydrous forms and its high procurement cost, which negatively impacts the overall economic feasibility of the process. The purification strategy depended extensively on silica gel column chromatography, a technique that generates massive volumes of solvent waste and limits batch sizes, making it unsuitable for the commercial scale-up of complex pharmaceutical intermediates. Additionally, the starting material synthesis involved violent exothermic steps that generated significant impurities, complicating downstream processing and reducing the overall yield of the high-purity OLED material or API precursor.
The Novel Approach
The innovative methodology presented in CN111094246B overcomes these historical barriers by implementing a fully thermal-driven process that utilizes standard jacketed reactors, ensuring consistent heat distribution and safety during exothermic events. By substituting hazardous condensing agents with safer alternatives like N,N'-carbonyldiimidazole (CDI), the process eliminates explosion risks and reduces raw material costs significantly. Crucially, the purification workflow has been redesigned to rely on crystallization and extraction techniques rather than chromatography, which drastically simplifies the isolation of the target compound and enhances environmental compliance. The introduction of specific protecting groups on the pyridine nucleus prevents unwanted side reactions, such as self-esterification, thereby improving the reaction selectivity and reducing the burden on downstream purification teams. This strategic redesign ensures stable amplification from kilogram to multi-ton scales without compromising the chemical integrity of the sensitive alkynyl functionality.
Mechanistic Insights into Pd-Catalyzed Sonogashira Coupling
The core transformation in this synthesis is the palladium-catalyzed cross-coupling between a protected bromopyridine intermediate and a substituted alkyne, a reaction that requires precise control over catalyst loading and base selection to minimize homocoupling byproducts. The patent specifies the use of bis(triphenylphosphine)palladium(II) dichloride in conjunction with cuprous iodide, a catalytic system that facilitates the oxidative addition and transmetallation steps necessary for carbon-carbon bond formation under mild thermal conditions. The presence of a hydroxyl protecting group, such as a methoxy or benzyl moiety, is mechanistically critical as it masks the nucleophilic phenolic oxygen, preventing it from interfering with the palladium cycle or undergoing undesired alkylation during the reaction. Solvent selection, particularly the use of tetrahydrofuran or N,N-dimethylacetamide, plays a vital role in solubilizing the organic substrates while maintaining the stability of the active catalytic species throughout the extended reaction times required for complete conversion. This careful orchestration of ligands, metals, and protecting groups ensures that the commercial scale-up of complex polymer additives or pharmaceutical scaffolds proceeds with high fidelity and minimal formation of difficult-to-remove metal residues.
Impurity control is further enhanced by the sequential deprotection strategy, where the carboxyl and hydroxyl groups are revealed in distinct stages to manage solubility and reactivity profiles. The use of lithium chloride as a reaction auxiliary during the deprotection step facilitates the cleavage of ether bonds under controlled conditions, avoiding the harsh acidic environments that could degrade the sensitive alkyne linkage. By optimizing the molar ratios of reaction auxiliaries to substrates, the process minimizes the formation of over-hydrolyzed byproducts or polymeric tars that often plague pyridine chemistry. The final hydrolysis step employs aqueous alkali under biphasic conditions, allowing for the efficient removal of ester protecting groups while keeping the organic product in a phase that can be easily separated from inorganic salts. This multi-stage approach to impurity management ensures that the final high-purity pharmaceutical intermediate meets the rigorous specifications demanded by downstream drug product manufacturers without requiring extensive reprocessing.
How to Synthesize Alkynyl Pyridine PHD Inhibitor Efficiently
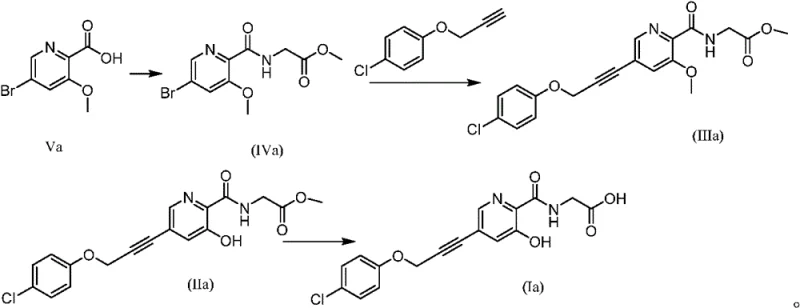
The synthesis of this target molecule involves a streamlined sequence beginning with the activation of the carboxylic acid followed by coupling and final deprotection. The detailed standardized synthesis steps see the guide below, which outlines the specific reagent grades, temperature ramps, and workup procedures necessary to replicate the patent's success in a GMP environment. Adhering to these parameters is essential for maintaining the reproducibility of the crystal form and ensuring that residual solvent levels remain within ICH guidelines. Operators must pay close attention to the addition rates of bases and the control of water content during the anhydrous coupling steps to prevent hydrolysis of the activated intermediates.
- Condensation of protected 3-substituted-5-bromopyridine-2-carboxylic acid with glycine ester using CDI.
- Palladium-catalyzed Sonogashira coupling with substituted alkyne under thermal conditions.
- Sequential removal of hydroxyl and carboxyl protecting groups using lithium chloride and alkaline hydrolysis.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this novel manufacturing route represents a significant opportunity to optimize the total cost of ownership for critical drug substances. By eliminating the reliance on specialized microwave equipment and silica gel chromatography, the process becomes compatible with existing multipurpose reactor infrastructure, thereby reducing capital expenditure requirements and increasing facility utilization rates. The removal of expensive and hazardous reagents like HOBT not only lowers direct material costs but also reduces the regulatory burden associated with handling explosive precursors, leading to lower insurance and compliance overheads. Furthermore, the shift to crystallization-based purification significantly decreases solvent consumption and waste disposal volumes, aligning with corporate sustainability goals and reducing the environmental footprint of the manufacturing campaign. These operational efficiencies translate into a more resilient supply chain capable of responding rapidly to fluctuating market demands without the bottlenecks associated with batch-limited purification technologies.
- Cost Reduction in Manufacturing: The substitution of costly condensing agents with economically viable alternatives like CDI directly lowers the bill of materials, while the avoidance of chromatographic purification reduces solvent purchase and recovery costs substantially. The ability to run reactions at standard thermal conditions rather than requiring energy-intensive microwave irradiation further decreases utility expenses per kilogram of product. Additionally, the improved yield profile resulting from better impurity control means that less starting material is required to produce the same amount of active pharmaceutical ingredient, compounding the savings across the entire production volume. These factors collectively drive a significant reduction in the cost of goods sold, making the final therapy more accessible while maintaining healthy margins for the manufacturer.
- Enhanced Supply Chain Reliability: Utilizing commodity chemicals and standard reactor types ensures that the supply chain is not vulnerable to shortages of specialized equipment or niche reagents that can disrupt production schedules. The robustness of the thermal coupling reaction allows for flexible batch sizing, enabling manufacturers to scale up or down based on real-time demand forecasts without requalifying the process. Moreover, the stability of the protected intermediates allows for potential stockpiling at strategic points in the synthesis, providing a buffer against upstream supply fluctuations. This flexibility is crucial for maintaining continuous supply to global partners, especially in the event of geopolitical disruptions or raw material scarcity affecting specific regions.
- Scalability and Environmental Compliance: The process design inherently supports green chemistry principles by minimizing waste generation through high-selectivity reactions and efficient isolation techniques. The elimination of silica gel waste streams simplifies the waste management protocol and reduces the hazard classification of the effluent, facilitating easier permitting and regulatory approval. Scalability is ensured by the linear relationship between heat transfer and reaction volume in standard vessels, allowing for seamless technology transfer from pilot plant to commercial manufacturing suites. This alignment with environmental, social, and governance (ESG) criteria enhances the corporate reputation of the supplier and meets the increasingly strict sustainability mandates of multinational pharmaceutical clients.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route in a regulated environment. These answers are derived directly from the experimental data and comparative examples provided in the patent documentation to ensure accuracy and relevance. Understanding these nuances is critical for technical teams evaluating the feasibility of adopting this new methodology for their specific portfolio of compounds.
Q: Why is the new method more suitable for industrial scale-up than the prior art?
A: The new method eliminates microwave irradiation and silica gel column chromatography, replacing them with standard thermal heating and crystallization, which are essential for ton-scale production.
Q: How does the process address safety concerns regarding condensing agents?
A: By utilizing N,N'-carbonyldiimidazole (CDI) instead of 1-hydroxybenzotriazole (HOBT), the process avoids the use of explosive anhydrous reagents, significantly enhancing operational safety.
Q: What is the impact of protecting groups on the overall impurity profile?
A: The use of specific hydroxyl and carboxyl protecting groups prevents unwanted self-esterification and side reactions during the coupling phase, leading to a cleaner crude product and higher purity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alkynyl Pyridine Prolyl Hydroxylase Inhibitor Supplier
At NINGBO INNO PHARMCHEM, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this sophisticated chemistry to life. Our technical team is adept at navigating the complexities of palladium-catalyzed couplings and protecting group manipulations, ensuring that your project benefits from our stringent purity specifications and rigorous QC labs. We understand that the transition from patent to plant requires more than just chemical knowledge; it demands a partnership focused on risk mitigation and timeline adherence. By leveraging our state-of-the-art facilities and deep process development expertise, we can help you secure a stable supply of this critical intermediate while optimizing the economic parameters of your supply chain.
We invite you to engage with our technical procurement team to discuss how we can tailor this manufacturing route to your specific volume and quality requirements. Request a Customized Cost-Saving Analysis today to understand the potential financial impact of switching to this scalable methodology. Our experts are ready to provide specific COA data and route feasibility assessments to support your internal decision-making processes. Let us help you overcome engineering bottlenecks and accelerate your path to market with a reliable partner committed to excellence in fine chemical manufacturing.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →