Advanced Synthesis of Forbitasvir RRRS Isomer: A Breakthrough in Chiral Pharmaceutical Intermediate Manufacturing
The pharmaceutical industry's relentless pursuit of effective Hepatitis C Virus (HCV) treatments has placed significant emphasis on the precise synthesis of NS5A inhibitors like Forbitasvir. Patent CN111205298A introduces a highly optimized preparation method for the Forbitasvir RRRS type isomer, a critical stereoisomer that dictates the medicinal efficacy of the final drug substance. This innovation addresses the complex challenge of constructing biaryl linkages while maintaining strict stereochemical control across four chiral centers. By leveraging a refined Suzuki-Miyaura cross-coupling strategy, the disclosed method offers a robust pathway for generating batch quantities of this high-value intermediate with superior purity profiles. For global procurement teams and R&D directors, this technology represents a pivotal shift towards more efficient and scalable manufacturing processes for complex antiviral agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for constructing the biaryl core of Forbitasvir often suffer from prolonged reaction times and incomplete conversions, which directly impact overall yield and production costs. Conventional protocols frequently rely on weaker or less soluble bases that necessitate extended heating periods, sometimes exceeding 16 hours, to achieve acceptable conversion rates. These extended thermal exposures increase the risk of side reactions and potential racemization at sensitive chiral centers, complicating downstream purification efforts. Furthermore, the use of suboptimal acid-binding agents can lead to significant accumulation of unreacted starting materials, such as the R-R type organic borate and R-S type aryl halide, requiring rigorous and costly chromatographic separation. The inefficiency of these legacy methods creates bottlenecks in supply chains, making it difficult to meet the demanding timelines required for clinical development and commercial launch.
The Novel Approach
The patented methodology revolutionizes this synthesis by identifying sodium carbonate as the superior acid-binding agent, drastically reducing the reflux time to merely 4 hours while ensuring near-complete consumption of reactants. This approach utilizes a specific solvent system comprising tetrahydrofuran, dimethylformamide, and water in a 5:1:1 ratio, which optimizes the solubility of both organic and inorganic components involved in the catalytic cycle. By controlling the molar ratio of sodium carbonate to the borate compound at 1.5:1, the process achieves a delicate balance that drives the equilibrium forward without introducing excessive inorganic salts that could complicate workup. The result is a streamlined operation that not only accelerates production throughput but also minimizes the formation of difficult-to-remove impurities. This efficiency translates directly into a more reliable pharmaceutical intermediate supplier capability, ensuring consistent quality and availability for downstream API manufacturing.
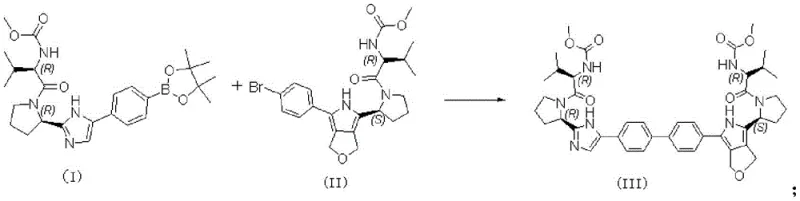
Mechanistic Insights into PdCl2(dppf)-Catalyzed Suzuki Coupling
The core of this synthetic breakthrough lies in the precise orchestration of the palladium-catalyzed cross-coupling between the R-R type organic borate ester and the R-S type aryl bromide. The catalyst system, utilizing PdCl2(dppf), facilitates the oxidative addition of the aryl halide to the palladium center, followed by transmetallation with the organoboron species activated by the carbonate base. The specific choice of ligands and the biphasic nature of the solvent system enhance the rate of reductive elimination, which is the turnover-limiting step in many Suzuki reactions involving sterically hindered substrates. This mechanistic efficiency allows the reaction to proceed rapidly at 90°C, avoiding the need for higher temperatures that could degrade the sensitive proline and macrocyclic motifs present in the molecular architecture. Understanding these kinetic parameters is essential for scaling the process from gram-scale laboratory experiments to multi-kilogram pilot runs without losing fidelity.
Controlling the impurity profile is paramount when synthesizing chiral intermediates, as even minor deviations can render the material unsuitable for pharmaceutical use. The optimized conditions prevent the hydrolysis of the boronic ester prior to coupling and minimize homocoupling side reactions that often plague Suzuki chemistries. By ensuring the reaction reaches completion within 4 hours, the process limits the exposure of the product to potentially degrading conditions, thereby preserving the integrity of the RRRS stereochemistry. The subsequent purification strategy, involving column chromatography followed by a specific crystallization step using methanol and sodium hydroxide, effectively removes trace palladium residues and any remaining achiral impurities. This rigorous control ensures the delivery of high-purity pharmaceutical intermediates that meet the stringent specifications required for regulatory filings and clinical trials.
How to Synthesize Forbitasvir RRRS Isomer Efficiently
The synthesis protocol outlined in the patent provides a clear roadmap for reproducing this high-efficiency coupling reaction in a controlled manufacturing environment. Operators must strictly adhere to the solvent ratios and the sequential addition of reagents to maintain the homogeneity of the reaction mixture throughout the reflux period. The detailed standard operating procedures ensure that the critical 1.5:1 molar equivalence of the base is maintained, which is the key variable determining the success of the transformation. For a comprehensive guide on the exact weighing, mixing, and isolation steps, please refer to the standardized synthesis instructions provided below.
- Prepare the solvent system by mixing tetrahydrofuran, dimethylformamide, and water in a 5: 1:1 volume ratio.
- Combine the R-R type organic borate compound and R-S type aryl halide with the solvent, adding PdCl2(dppf) catalyst and sodium carbonate base.
- Reflux the mixture at 90°C for 4 hours under nitrogen, then purify the crude product via column chromatography and crystallization.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this optimized synthesis route offers profound economic and logistical benefits for organizations managing the supply chain of antiviral therapeutics. The reduction in reaction time from over 16 hours to just 4 hours significantly increases the throughput of existing reactor vessels, allowing manufacturers to produce larger batches within the same operational window. This efficiency gain directly contributes to cost reduction in API manufacturing by lowering energy consumption and maximizing asset utilization without requiring capital investment in new equipment. Furthermore, the use of inexpensive and readily available sodium carbonate replaces costlier or less efficient organic bases, simplifying the raw material sourcing strategy and reducing overall material costs.
- Cost Reduction in Manufacturing: The elimination of prolonged heating cycles and the use of a cheap inorganic base dramatically lower the variable costs associated with each production batch. By avoiding the need for exotic reagents or complex cryogenic conditions, the process becomes inherently more economical and easier to budget for long-term production contracts. The simplified workup procedure also reduces the consumption of solvents and silica gel during purification, further driving down the cost of goods sold. These cumulative savings make the final intermediate more price-competitive in the global market while maintaining healthy margins for the manufacturer.
- Enhanced Supply Chain Reliability: Shorter cycle times mean that production schedules become more flexible and responsive to sudden changes in demand from downstream API clients. The robustness of the reaction conditions reduces the risk of batch failures due to minor process deviations, ensuring a steady and predictable flow of material. This reliability is crucial for reducing lead time for high-purity intermediates, allowing pharmaceutical companies to accelerate their own formulation and testing timelines. A stable supply of this key building block mitigates the risk of drug shortages and supports continuous commercial manufacturing operations.
- Scalability and Environmental Compliance: The process is designed with commercial scale-up of complex chiral molecules in mind, utilizing standard equipment and conditions that translate easily from the lab to the plant. The aqueous workup and reduced solvent usage align with green chemistry principles, minimizing the environmental footprint and simplifying waste disposal compliance. The ability to scale without re-optimizing critical parameters ensures that the quality established at the small scale is preserved at the metric-ton level. This scalability assures partners that the supply can grow in lockstep with the commercial success of the final hepatitis C medication.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this specific isomer. These answers are derived directly from the experimental data and claims presented in the patent documentation to ensure accuracy and relevance. Understanding these details helps stakeholders make informed decisions about integrating this intermediate into their development pipelines.
Q: Why is sodium carbonate preferred over other bases for this Suzuki coupling?
A: Sodium carbonate significantly reduces reaction time to just 4 hours compared to 16 hours with potassium acetate, ensuring more complete conversion and lower residual starting materials.
Q: What is the critical molar ratio for the base in this synthesis?
A: The patent specifies a molar ratio of sodium carbonate to the R-R type organic borate compound of 1.5:1 as optimal for driving the reaction to completion without excess waste.
Q: How is the stereochemical integrity maintained during the coupling?
A: The mild conditions (90°C reflux) and specific catalyst system (PdCl2(dppf)) prevent racemization of the chiral centers present in the R-R and R-S precursors.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Forbitasvir RRRS Isomer Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving antiviral therapies. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements regardless of the project stage. We are committed to delivering materials with stringent purity specifications, supported by our rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify every batch. Our expertise in chiral synthesis and cross-coupling reactions positions us as an ideal partner for navigating the complexities of modern pharmaceutical manufacturing.
We invite you to contact our technical procurement team to discuss how we can support your specific project needs with tailored solutions. Request a Customized Cost-Saving Analysis today to understand how our optimized processes can benefit your bottom line. We are ready to provide specific COA data and route feasibility assessments to demonstrate our capability to be your trusted long-term source for this vital intermediate.