Advanced Synthetic Route for N4-Acyl-5'-Deoxy-5-Fluorocytidine Derivatives: Enhancing Purity and Commercial Scalability
The pharmaceutical industry continuously seeks robust and efficient synthetic pathways for critical anticancer agents, particularly for the production of capecitabine and its precursors. Patent CN100569790C, granted in late 2009, introduces a groundbreaking methodology for synthesizing N4-acyl-5'-deoxy-5-fluorocytidine derivatives, which serve as vital intermediates in the manufacture of oral fluoropyrimidine carbamate antineoplastic agents. This intellectual property outlines a streamlined three-step reaction sequence that begins with 5'-deoxy-5-fluorocytidine (Compound II) and proceeds through two novel key intermediates, Compound III and Compound IV, before yielding the final target Compound I. The innovation lies in the strategic application of bis(trichloromethyl)carbonate, commonly known as triphosgene, to introduce a protective cyclic lactone onto the sugar moiety's hydroxyl groups. This approach not only simplifies the synthetic tree but also addresses long-standing challenges regarding yield optimization and operational efficiency in nucleoside chemistry. For global supply chain leaders and R&D directors, understanding this patented route is essential for evaluating potential partnerships with a reliable pharmaceutical intermediate supplier capable of delivering high-purity materials.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art technologies, such as those described in European Patent EP 0316704A and Chinese Patent CN1035675C, have historically relied on cumbersome multi-step protection strategies to synthesize these valuable nucleoside derivatives. These conventional methods typically necessitate the sequential introduction of various protecting groups, including acyl, isopropylidene, and silyl moieties, to mask the reactive hydroxyl functions on the ribose sugar before the N4-amino group can be acylated. This lengthy sequence inherently extends the production cycle time and increases the complexity of the manufacturing process. Furthermore, a significant drawback of these older methodologies is the propensity for side reactions, particularly the inadvertent removal of the acyl group from the amino position during the deprotection phases. Such instability necessitates complicated separation and purification processes to isolate the desired product, leading to substantial material loss and increased operational costs. Additionally, alternative methods like those in CN1053194C attempted to simplify the process by using identical protecting groups for both hydroxyl and amino functions, but this often required the mass consumption of costly acylating agents, thereby negating any potential economic benefits and failing to provide a truly cost-effective solution for cost reduction in pharmaceutical intermediate manufacturing.
The Novel Approach
In stark contrast to the convoluted pathways of the past, the method disclosed in CN100569790C offers a remarkably concise and efficient three-step synthesis that dramatically reduces processing time and resource consumption. The core innovation involves the use of triphosgene as a highly selective acylating agent to form a cyclic carbonate bridge between the 2' and 3' hydroxyl groups of the sugar moiety in a single operation. This protective strategy effectively blocks the hydroxyls from participating in subsequent reactions while leaving the N4-amino group available for selective functionalization. By bypassing the need for multiple distinct protecting groups and their associated removal steps, this novel approach minimizes the number of unit operations required, thereby enhancing the overall throughput of the manufacturing line. The process utilizes readily available raw materials and standard organic solvents, making it highly amenable to scale-up. As illustrated in the reaction scheme below, the transformation from the starting material to the final product is direct and logical, avoiding the pitfalls of previous techniques.
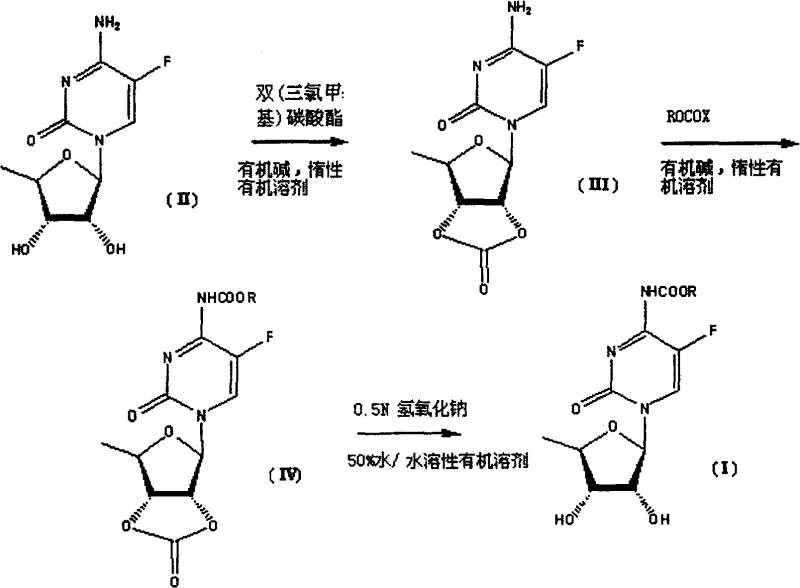
Mechanistic Insights into Triphosgene-Mediated Protection and Selective Acylation
The success of this synthetic route hinges on the unique reactivity of bis(trichloromethyl)carbonate (triphosgene) in the presence of organic bases such as pyridine or triethylamine. In the first step, triphosgene reacts with the vicinal diol system of the 5'-deoxy-5-fluorocytidine sugar moiety to form a five-membered cyclic carbonate ring, resulting in Compound III. This cyclization is thermodynamically favorable and kinetically rapid under mild conditions, typically between 20°C and 50°C. The formation of this rigid cyclic structure not only protects the hydroxyl groups but also locks the sugar conformation, which can influence the stereochemical outcome of subsequent reactions. The intermediate Compound III is a stable entity that can be isolated or carried forward directly, providing flexibility in process design. The use of triphosgene is particularly advantageous because it acts as a safe and solid equivalent of phosgene, allowing for precise stoichiometric control and safer handling in a commercial plant environment compared to gaseous reagents.
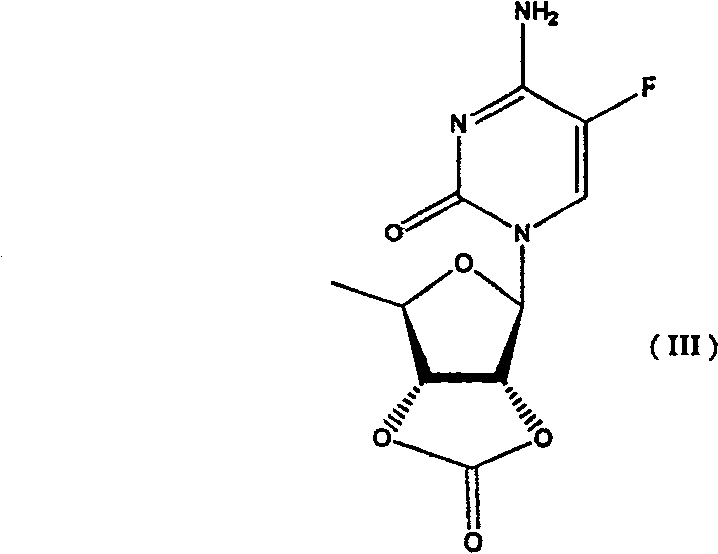
Following the protection step, the N4-amino group undergoes acylation with an activated carboxylic acid derivative (ROCOX), such as an acid chloride or anhydride, to form Compound IV. The presence of the cyclic carbonate ensures that acylation occurs exclusively at the nitrogen atom, preventing the formation of O-acyl impurities. The final and perhaps most critical step is the selective hydrolysis of the cyclic carbonate in Compound IV to regenerate the free 2',3'-diol system of the final product, Compound I. This is achieved using a dilute mineral alkali, such as 0.5M sodium hydroxide, in a mixed solvent system of water and a water-miscible organic solvent like dioxane or ethanol. The conditions are carefully tuned to be mild enough (0°C to 10°C) to hydrolyze the carbonate ester without cleaving the more stable N4-acyl amide bond. This chemoselectivity is the key to the high purity and yield of the final product, ensuring that the valuable N4-substituent remains intact while the temporary protecting group is cleanly removed.
How to Synthesize N4-Acyl-5'-Deoxy-5-Fluorocytidine Efficiently
The execution of this synthesis requires careful attention to reaction parameters, particularly temperature control and stoichiometry, to maximize the yield of the key intermediates. The process begins with the dissolution of the starting nucleoside in an anhydrous halogenated solvent, followed by the addition of a tertiary amine base to scavenge the hydrogen chloride generated during the reaction with triphosgene. Once the cyclic carbonate intermediate is secured, the acylation step is performed at lower temperatures to prevent degradation, utilizing an excess of the acylating agent to drive the reaction to completion. The final hydrolysis step demands precise pH control and monitoring to ensure complete deprotection without over-hydrolysis of the product. For detailed operational protocols, safety guidelines, and specific workup procedures, please refer to the standardized synthesis guide below.
- React 5'-deoxy-5-fluorocytidine with bis(trichloromethyl)carbonate (triphosgene) and an organic base in an inert solvent to form the protected cyclic carbonate intermediate (Compound III).
- Perform N4-acylation on Compound III using an acylating agent (ROCOX) in the presence of an organic base to generate the N-protected intermediate (Compound IV).
- Execute selective hydrolysis of the cyclic carbonate moiety in Compound IV using dilute mineral alkali in a water-miscible organic solvent mixture to obtain the final product (Compound I).
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this triphosgene-mediated synthetic route offers profound advantages for procurement managers and supply chain heads looking to optimize their sourcing strategies for anticancer intermediates. The elimination of multiple protection and deprotection steps translates directly into a significantly reduced manufacturing footprint and lower utility consumption. By shortening the synthetic sequence from potentially six or more steps down to just three, the overall process time is drastically compressed, allowing for faster turnover of production batches and improved responsiveness to market demand. Furthermore, the use of triphosgene, a commodity chemical, replaces the need for specialized and expensive silylating agents or complex anhydrides often required in traditional routes, leading to substantial cost savings in raw material procurement. The simplified purification requirements, primarily involving extraction and crystallization rather than complex chromatography, further reduce the cost of goods sold (COGS) and minimize solvent waste generation.
- Cost Reduction in Manufacturing: The streamlined nature of this process inherently lowers production costs by reducing the number of unit operations and the quantity of reagents consumed. The avoidance of expensive protecting group chemistry and the ability to use cheaper acylating agents for the N4-position contribute to a more economical process. Additionally, the high selectivity of the reactions minimizes the formation of difficult-to-remove impurities, which reduces the burden on downstream purification and increases the overall recovery of the final product. This efficiency gain allows suppliers to offer more competitive pricing structures for high-purity pharmaceutical intermediates without compromising on quality standards.
- Enhanced Supply Chain Reliability: The reliance on widely available starting materials like 5'-deoxy-5-fluorocytidine and common solvents such as methylene chloride and pyridine ensures a robust and resilient supply chain. Unlike processes that depend on exotic catalysts or hard-to-source reagents, this method mitigates the risk of supply disruptions. The stability of the intermediates, particularly the cyclic carbonate Compound III, allows for potential stockpiling or flexible scheduling of the subsequent acylation step. This flexibility is crucial for maintaining continuous production schedules and meeting the rigorous delivery timelines expected by global pharmaceutical clients.
- Scalability and Environmental Compliance: The reaction conditions employed in this patent are mild and operate within a safe temperature range, making the process highly scalable from laboratory benchtop to multi-ton commercial reactors. The use of standard extraction and crystallization techniques for isolation facilitates easy technology transfer to large-scale manufacturing facilities. Moreover, the reduction in the number of chemical steps and the use of recyclable solvents contribute to a smaller environmental footprint. The process generates less hazardous waste compared to traditional methods involving heavy metals or toxic silyl reagents, aligning with modern green chemistry principles and stringent environmental regulations.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of this synthetic route for their specific applications, we have compiled a set of frequently asked questions based on the technical disclosures within the patent. These inquiries address common concerns regarding reaction selectivity, scalability, and the comparative advantages of this method over legacy technologies. Understanding these nuances is critical for R&D directors assessing the potential for process integration and for quality assurance teams verifying the purity profiles of the resulting intermediates. The answers provided below are derived directly from the experimental data and theoretical framework presented in the intellectual property documentation.
Q: What are the primary advantages of using triphosgene in this synthesis compared to traditional methods?
A: The use of bis(trichloromethyl)carbonate (triphosgene) allows for the simultaneous protection of the 2' and 3' hydroxyl groups as a cyclic carbonate in a single step. This eliminates the need for multiple protection and deprotection sequences involving silyl or isopropylidene groups found in older patents, significantly reducing reaction time, simplifying purification, and lowering the overall consumption of expensive reagents.
Q: How does this novel route address the issue of side reactions during acylation?
A: By introducing the cyclic carbonate protective group first, the hydroxyl groups on the sugar moiety are effectively blocked, preventing unwanted O-acylation. This ensures that the subsequent acylation step occurs selectively at the N4-amino position. Furthermore, the mild hydrolysis conditions used in the final step specifically target the cyclic carbonate without cleaving the newly formed N4-acyl bond, thereby minimizing side products and improving the purity profile of the final intermediate.
Q: Is this synthetic method suitable for large-scale industrial manufacturing?
A: Yes, the process is explicitly designed for industrial scalability. It utilizes readily available starting materials like 5'-deoxy-5-fluorocytidine and common solvents such as methylene chloride and pyridine. The reaction conditions are mild (ranging from 0°C to 60°C), and the workup procedures involve standard extraction and crystallization techniques, making it highly adaptable for commercial scale-up from pilot plants to multi-ton production facilities.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N4-Acyl-5'-Deoxy-5-Fluorocytidine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development and commercialization of life-saving oncology drugs. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in patents like CN100569790C can be translated into reliable, large-scale supply. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of N4-acyl-5'-deoxy-5-fluorocytidine meets the exacting standards required for GMP manufacturing. Our commitment to technical excellence allows us to navigate the complexities of nucleoside chemistry, delivering materials that support your clinical and commercial goals with consistency and precision.
We invite you to engage with our technical procurement team to discuss how our advanced manufacturing capabilities can support your specific project requirements. Whether you are in the early stages of process development or looking to secure a long-term supply for commercial launch, we are prepared to provide a Customized Cost-Saving Analysis tailored to your volume needs. Please contact us today to request specific COA data, route feasibility assessments, and samples to evaluate the superior quality of our intermediates. Let us partner with you to accelerate your drug development timeline through superior chemistry and supply chain reliability.