Scalable Synthesis of N-(Benzyloxycarbonyl)-amino-epoxy-phenylbutane for HIV Protease Inhibitors
The pharmaceutical industry's relentless pursuit of effective antiretroviral therapies has placed significant emphasis on the efficient production of key intermediates for HIV protease inhibitors. Patent CN101486691B introduces a robust and highly efficient methodology for synthesizing N-(benzyloxycarbonyl)-amino-epoxy-phenylbutane, a critical building block for drugs such as Saquinavir and Nelfinavir. This technical disclosure represents a paradigm shift from traditional, low-yield synthetic routes that relied heavily on cumbersome purification techniques. By leveraging L-phenylalanine as a chiral starting material, the described process achieves a remarkable total molar yield of 75% and an optical purity exceeding 99%, addressing the longstanding challenges of cost and scalability in antiretroviral drug manufacturing. For R&D directors and supply chain strategists, this patent offers a compelling blueprint for optimizing the production of complex chiral intermediates without compromising on quality or safety standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of epoxy-phenylbutane derivatives has been plagued by inefficient processes that hindered commercial viability. Prior art, such as the method disclosed in EP604185A1, necessitated a series of three rapid flash chromatography separations followed by recrystallization at cryogenic temperatures of -78°C using dichloromethane. This approach not only resulted in a dismal yield of merely 26.2% but also imposed severe operational burdens regarding solvent consumption, energy usage for deep cooling, and the handling of hazardous halogenated solvents on a large scale. Furthermore, other documented routes found in patents like WO2002064553A1 and WO9845271A1 often involved excessively long reaction sequences or the liberal use of highly flammable and explosive chemicals, creating substantial safety risks and supply chain vulnerabilities for manufacturers aiming to produce these vital HIV medication components reliably.
The Novel Approach
In stark contrast, the methodology outlined in CN101486691B streamlines the synthetic pathway into four distinct, high-efficiency steps that eliminate the need for chromatographic purification entirely. By initiating the synthesis with L-phenylalanine, a readily available and cost-effective amino acid, the process inherently embeds the necessary stereochemistry from the outset. The novel route employs a controlled diazotization followed by selective reduction and hydrolysis, culminating in a base-mediated cyclization. This strategic design allows for the isolation of intermediates through simple crystallization and washing procedures rather than complex column chromatography. The result is a process that not only triples the overall yield compared to legacy methods but also significantly enhances operational safety by minimizing the inventory of unstable intermediates and avoiding the need for extreme cryogenic conditions during the final purification stages.
Mechanistic Insights into Chiral Pool Synthesis and Epoxide Formation
The core of this synthetic strategy lies in the precise manipulation of the chiral center derived from L-phenylalanine to ensure the final epoxide retains the required (S,S) configuration essential for biological activity. The initial step involves the activation of the carboxylic acid group of the protected phenylalanine derivative using an acid activator such as ethyl chloroformate in the presence of pyridine. This forms a mixed anhydride intermediate which is subsequently reacted with diazomethane to generate the corresponding diazoketone. This transformation is critical as it extends the carbon chain while preserving the alpha-chirality of the amino acid backbone. The reaction is conducted at controlled low temperatures between -60°C and -20°C to mitigate the risk of racemization and to manage the exothermic nature of the diazotization, ensuring that the stereochemical integrity established by the natural amino acid source is not compromised before the subsequent functionalization steps.
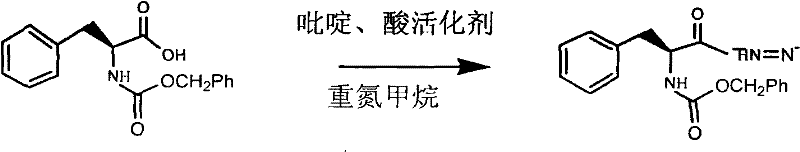
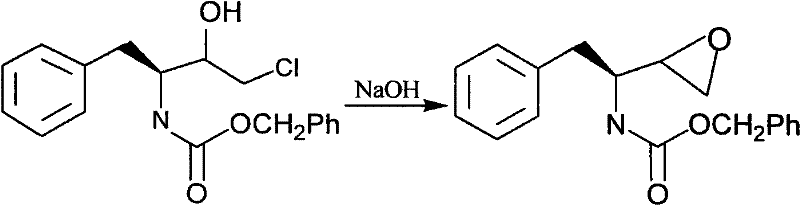
Following the formation of the diazoketone, the process proceeds to a selective reduction and chlorination phase. The diazoketone is treated with a solution of hydrogen chloride in ethyl acetate at temperatures ranging from -5°C to -40°C. This step effectively converts the diazo group into a chloromethyl ketone functionality. The mechanistic pathway here involves the protonation of the diazo group followed by nucleophilic attack by the chloride ion, displacing nitrogen gas. This transformation is pivotal as it installs the leaving group necessary for the eventual ring closure. The subsequent reduction of the ketone to a chlorohydrin using catalysts like aluminum isopropoxide or sodium borohydride is performed with high stereoselectivity. The choice of reducing agent and solvent system is optimized to favor the formation of the specific diastereomer required for the final epoxide, minimizing the formation of unwanted isomers that would otherwise lower the optical purity of the final API intermediate.

The final stage of the synthesis involves the intramolecular cyclization of the chlorohydrin intermediate to form the strained epoxide ring. This is achieved by treating the crude chlorohydrin with an inorganic base, such as sodium hydroxide, in a biphasic system containing water and an organic solvent like acetone or ethyl acetate. The base deprotonates the hydroxyl group of the chlorohydrin, generating an alkoxide ion which then performs an intramolecular SN2 attack on the adjacent carbon bearing the chlorine atom. This displacement reaction closes the three-membered epoxide ring with inversion of configuration at the carbon center, locking in the desired stereochemistry. The use of mild basic conditions and moderate temperatures (20°C to 40°C) ensures that the sensitive epoxide ring is formed without undergoing hydrolysis or polymerization, resulting in a final product with an optical purity of greater than 99% and a high degree of chemical purity suitable for downstream coupling reactions in the synthesis of Saquinavir.
How to Synthesize N-(Benzyloxycarbonyl)-amino-epoxy-phenylbutane Efficiently
The synthesis of this high-value intermediate requires strict adherence to anhydrous conditions during the initial steps to prevent premature hydrolysis of the activated intermediates. The process begins with the dissolution of L-phenylalanine in a suitable organic solvent such as ethyl acetate, followed by cooling to sub-zero temperatures before the addition of activators. Detailed standard operating procedures regarding stoichiometry, temperature ramps, and workup protocols are essential to replicate the high yields reported in the patent literature. For process chemists looking to implement this route, understanding the kinetics of the diazomethane addition and the precise control of pH during the final cyclization are critical parameters for success. The detailed standardized synthesis steps are provided in the guide below.
- Diazotization of L-Phenylalanine derivative using pyridine and acid activator followed by reaction with diazomethane to form the diazoketone intermediate.
- Selective reduction and chlorination using hydrogen chloride in ethyl acetate at low temperatures (-5 to -40°C) to generate the chloro-ketone species.
- Final cyclization using inorganic base in organic solvent to close the epoxide ring, yielding the target product with >99% optical purity.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this synthetic route offers transformative economic and logistical benefits over traditional methods. The elimination of flash chromatography, which is notoriously difficult to scale and solvent-intensive, translates directly into substantial reductions in manufacturing costs and processing time. By relying on crystallization and distillation for purification, the process becomes amenable to large-scale batch production in standard stainless steel reactors, removing the bottleneck associated with preparative HPLC or silica gel columns. This shift not only lowers the cost of goods sold (COGS) but also drastically reduces the environmental footprint of the manufacturing process by minimizing solvent waste generation, aligning with modern green chemistry initiatives and regulatory compliance requirements for pharmaceutical suppliers.
- Cost Reduction in Manufacturing: The most significant economic driver of this technology is the tripling of the overall process yield from approximately 26% in legacy methods to 75% in this patented route. This dramatic increase in efficiency means that for every kilogram of final product, significantly less raw material is consumed, directly lowering the variable cost of production. Furthermore, the avoidance of expensive chromatography resins and the reduction in solvent volume required for purification lead to considerable savings in utility and waste disposal costs. The use of L-phenylalanine, a commodity fermentation product, as the starting material ensures a stable and predictable raw material cost base, shielding the supply chain from the volatility often associated with specialized synthetic building blocks.
- Enhanced Supply Chain Reliability: The robustness of this synthetic pathway enhances supply chain continuity by reducing the number of critical process steps that could potentially fail or require rework. Traditional methods involving multiple chromatographic separations are prone to variability and batch failures, which can disrupt delivery schedules for downstream API manufacturers. In contrast, this crystallization-based process offers high reproducibility and consistent quality, ensuring that delivery timelines are met with greater certainty. Additionally, the reliance on common chemical reagents and solvents such as ethyl acetate, toluene, and sodium hydroxide mitigates the risk of supply shortages for exotic or regulated chemicals, further securing the production pipeline against external market disruptions.
- Scalability and Environmental Compliance: The process is inherently designed for commercial scale-up, utilizing unit operations that are standard in the fine chemical industry. The ability to perform the reaction in relatively concentrated solutions and the ease of isolating intermediates via filtration or phase separation facilitate the transition from pilot plant to multi-ton production without the need for specialized equipment. From an environmental standpoint, the reduction in solvent usage and the avoidance of halogenated solvents in the final purification steps simplify waste treatment and reduce the burden on effluent processing facilities. This alignment with environmental, social, and governance (ESG) goals makes the technology attractive for multinational corporations seeking to minimize the ecological impact of their pharmaceutical supply chains.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and process descriptions found in the patent documentation, providing clarity on the feasibility and advantages of the method for potential partners and licensees. Understanding these details is crucial for evaluating the technology's fit within existing manufacturing frameworks.
Q: What is the primary advantage of this synthesis route over prior art like EP604185A1?
A: The primary advantage is the drastic improvement in yield and process simplicity. While prior art methods required three flash chromatography steps and cryogenic recrystallization at -78°C resulting in only 26.2% yield, this patented method achieves a total molar yield of 75% using standard crystallization techniques, significantly reducing production costs and time.
Q: How is chiral integrity maintained during the synthesis?
A: Chiral integrity is maintained by utilizing L-Phenylalanine as the starting chiral pool material. The reaction conditions, particularly the low-temperature diazotization and controlled reduction steps, are optimized to prevent racemization, ensuring the final product maintains an optical purity of greater than 99%.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process is specifically designed for industrial scalability. It avoids complex purification methods like column chromatography, relies on readily available starting materials like L-Phenylalanine, and utilizes standard unit operations such as distillation and crystallization, making it highly viable for metric-ton scale production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-(Benzyloxycarbonyl)-amino-epoxy-phenylbutane Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the global fight against HIV. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in CN101486691B can be translated into reliable industrial reality. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to guarantee that every batch of N-(benzyloxycarbonyl)-amino-epoxy-phenylbutane meets the exacting standards required for GMP API synthesis. Our commitment to quality assurance ensures that our clients receive materials that facilitate smooth downstream processing and consistent final drug product quality.
We invite pharmaceutical companies and contract manufacturing organizations to collaborate with us to leverage this cost-effective and scalable synthetic route. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and production timelines. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that optimize your supply chain and reduce the overall cost of your antiretroviral drug manufacturing programs.