Revolutionizing Difluoromethyl Oxazolidinone Production via Safe Two-Step Catalytic Fluorination
Revolutionizing Difluoromethyl Oxazolidinone Production via Safe Two-Step Catalytic Fluorination
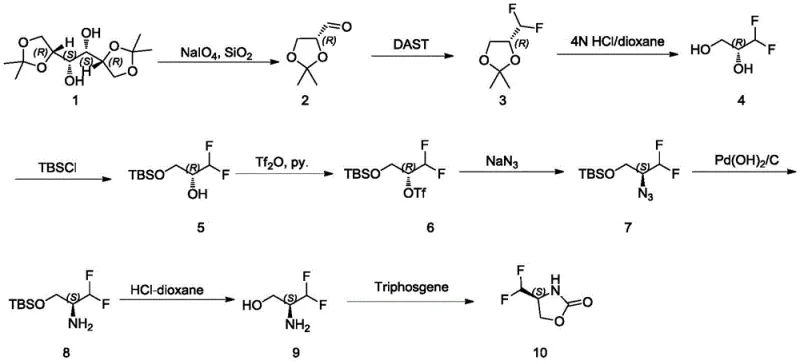
The pharmaceutical industry continuously seeks robust synthetic pathways that balance high purity with operational safety, particularly for fluorinated intermediates used in kinase inhibitor development. Patent CN109265408B introduces a groundbreaking methodology for synthesizing difluoromethyl substituted dioxane-2-ketone derivatives, specifically targeting the critical 4,4-(difluoromethyl)oxazolidine-2-one scaffold. This innovation addresses significant bottlenecks found in legacy processes, such as the reliance on unstable intermediates and hazardous reagents like sodium azide. By condensing a previously nine-step sequence into a concise two-step operation involving direct fluorination and subsequent deprotection, this technology offers a compelling value proposition for manufacturers of high-purity pharmaceutical intermediates. The strategic shift not only enhances atom economy but also drastically simplifies the supply chain logistics required for producing these complex heterocyclic building blocks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 4,4-(difluoromethyl)oxazolidine-2-ketone has been plagued by severe operational inefficiencies and safety hazards, as detailed in prior art such as WO2017001645A1. The traditional route necessitates a cumbersome nine-step sequence involving oxidation, fluorination, multiple protection and deprotection cycles, and a dangerous azide substitution step. A critical failure point in this legacy workflow is the instability of the fluorinated aldehyde intermediate, which degrades rapidly even under inert argon atmospheres at low temperatures, leading to significant material loss. Furthermore, the fluorination reaction in the old process is notoriously difficult to control; if the precursor purity drops below 80%, the reaction becomes chaotic, yielding less than 30% of the desired product after purification. The reliance on sodium azide introduces unacceptable explosion risks, while the subsequent hydrogenation requires expensive palladium catalysts, creating a cost structure that is unsustainable for large-scale commercial operations.
The Novel Approach
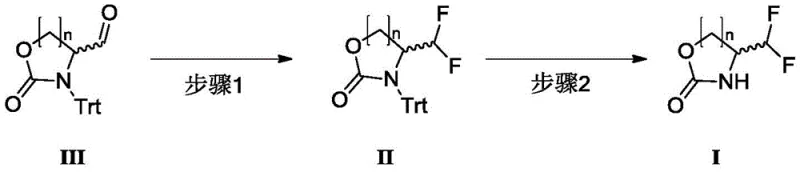
In stark contrast to the convoluted legacy workflows, the methodology disclosed in CN109265408B utilizes a direct and efficient two-step strategy starting from a stable trityl-protected aldehyde precursor. The first step involves the fluorination of the aldehyde group using reagents like DAST or BAST under mild conditions, directly installing the difluoromethyl motif with high fidelity. This is immediately followed by a straightforward deprotection step using hydrochloric acid to remove the trityl protecting group, yielding the final oxazolidinone product. This streamlined approach completely bypasses the need for azide chemistry and precious metal catalysis, thereby eliminating major safety liabilities. The process is characterized by excellent controllability, allowing for real-time monitoring via simple phosphomolybdic acid staining, which ensures consistent quality and minimizes the formation of by-products such as monofluoro species. This represents a paradigm shift towards greener, safer, and more economically viable manufacturing of fluorinated heterocycles.
Mechanistic Insights into DAST-Mediated Fluorination and Acidic Deprotection
The core chemical transformation in this novel pathway relies on the nucleophilic fluorination capability of diethylaminosulfur trifluoride (DAST) or its analogues like BAST. In the first step, the carbonyl oxygen of the trityl-protected oxazolidine-4-carbaldehyde attacks the sulfur center of the fluorinating agent, forming a reactive intermediate that facilitates the replacement of the carbonyl oxygen with two fluorine atoms. This mechanism proceeds efficiently at temperatures between 0°C and 35°C, avoiding the thermal degradation issues seen in previous methods. The reaction is highly selective, preventing the formation of ectopic difluoro isomers or monofluoro impurities, which is crucial for maintaining the integrity of the chiral center at the 4-position of the oxazolidine ring. The use of solvents such as dichloromethane or toluene further optimizes the reaction kinetics, ensuring complete conversion while maintaining a homogeneous reaction mixture that is easy to handle.
Following fluorination, the second mechanistic phase involves the cleavage of the trityl (triphenylmethyl) protecting group from the nitrogen atom of the oxazolidine ring. This deprotection is achieved through acidolysis using hydrochloric acid in dioxane or hydrogen chloride gas. The acidic conditions protonate the nitrogen or facilitate the formation of a stable trityl cation, which dissociates from the molecule to reveal the free NH group of the final product. This step is remarkably robust, functioning effectively across a temperature range of 0°C to 50°C without compromising the sensitive difluoromethyl group. The simplicity of this acid-catalyzed cleavage allows for easy workup procedures, typically involving concentration and column chromatography, resulting in a final product with purity levels reaching 99%. This mechanistic elegance ensures that the stereochemical configuration, whether (S) or (R), is preserved throughout the synthesis.
How to Synthesize 4-(Difluoromethyl)oxazolidin-2-one Efficiently
The implementation of this synthesis route requires precise control over reaction parameters to maximize yield and safety, yet the operational complexity is significantly lower than traditional methods. The process begins with the dissolution of the trityl-protected aldehyde in an appropriate organic solvent, followed by the controlled addition of the fluorinating agent under inert atmosphere to manage exothermicity. After the fluorination is complete and quenched safely, the intermediate is isolated and subjected to acidic conditions to remove the protecting group. Detailed standard operating procedures regarding stoichiometry, specific solvent grades, and quenching protocols are essential for reproducibility. For a comprehensive guide on the exact experimental conditions and workup procedures validated in the patent examples, please refer to the standardized synthesis steps outlined below.
- Step 1: Fluorinate the trityl-protected oxazolidine-4-carbaldehyde (Compound III) using DAST or BAST at 0-35°C to form the difluoromethyl intermediate (Compound II).
- Step 2: Perform deprotection on Compound II using hydrochloric acid in dioxane or hydrogen chloride gas at 0-50°C to remove the trityl group and yield the final product (Compound I).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis method translates into tangible strategic benefits regarding cost stability and risk mitigation. By eliminating the need for sodium azide, facilities can avoid the stringent regulatory compliance and specialized storage infrastructure associated with explosive precursors, thereby lowering overhead costs related to safety management. Furthermore, the removal of expensive palladium catalysts from the process flow reduces the dependency on volatile precious metal markets and eliminates the costly downstream processing steps required to reduce residual heavy metals to ppm levels. The simplified two-step nature of the reaction also implies a shorter production cycle time, which enhances the responsiveness of the supply chain to fluctuating market demands for kinase inhibitor intermediates.
- Cost Reduction in Manufacturing: The economic advantages of this process are driven primarily by the drastic reduction in unit operations and the elimination of high-cost reagents. By condensing nine steps into two, the cumulative yield loss inherent in multi-step synthesis is minimized, leading to a substantial increase in overall mass efficiency. The avoidance of palladium on carbon and sodium azide removes two of the most expensive line items from the bill of materials, while the use of commodity chemicals like hydrochloric acid and DAST ensures predictable pricing. Additionally, the simplified purification process reduces solvent consumption and waste disposal costs, contributing to a leaner manufacturing cost structure that supports competitive pricing for the final API intermediate.
- Enhanced Supply Chain Reliability: Supply continuity is significantly bolstered by the robustness of the reaction conditions and the stability of the intermediates involved. Unlike the legacy process where intermediates degraded within days, the compounds in this new route are stable enough for standard storage and transport, reducing the risk of batch failures due to material degradation. The ability to monitor the reaction progress using simple thin-layer chromatography with phosphomolybdic acid staining empowers quality control teams to make real-time decisions, preventing the propagation of off-spec material through the production line. This reliability ensures that delivery schedules can be met consistently, a critical factor for pharmaceutical clients managing tight development timelines.
- Scalability and Environmental Compliance: From an environmental and scale-up perspective, this methodology offers a cleaner profile that aligns with modern green chemistry principles. The high atom economy and the absence of toxic azide waste streams simplify effluent treatment and reduce the environmental footprint of the manufacturing site. The process has already been demonstrated to be scalable to the hectogram level with high yields, indicating a clear path to ton-scale production without encountering unforeseen engineering challenges. This scalability ensures that the supplier can grow alongside the client's needs, from clinical trial quantities to commercial launch volumes, without requiring major process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of difluoromethyl substituted oxazolidinones. These insights are derived directly from the experimental data and comparative analysis presented in the patent literature, providing a transparent view of the technology's capabilities. Understanding these details helps stakeholders assess the feasibility of integrating this intermediate into their own drug discovery pipelines.
Q: How does this new method improve safety compared to traditional synthesis routes?
A: The novel process eliminates the use of highly hazardous sodium azide and expensive palladium catalysts required in previous 9-step methods, significantly reducing explosion risks and heavy metal contamination concerns.
Q: What represents the key advantage in terms of reaction monitoring and yield?
A: Unlike prior art where intermediates were unstable and hard to detect, this method allows easy monitoring via phosphomolybdic acid staining and achieves a combined two-step yield of up to 82% with 99% purity.
Q: Is this synthesis method suitable for large-scale industrial manufacturing?
A: Yes, the process has been successfully demonstrated at the hectogram level with simple post-treatment procedures, making it highly scalable for commercial production without complex purification steps.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-(Difluoromethyl)oxazolidin-2-one Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality fluorinated intermediates play in the development of next-generation therapeutics. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless. We adhere to stringent purity specifications and utilize rigorous QC labs to guarantee that every batch of 4-(difluoromethyl)oxazolidin-2-one meets the exacting standards required for pharmaceutical applications. Our commitment to process safety and efficiency mirrors the innovations found in CN109265408B, allowing us to offer a product that is both cost-effective and reliable.
We invite potential partners to engage with our technical procurement team to discuss how this advanced synthesis route can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits specific to your volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, ensuring that your project moves forward with the highest quality building blocks available in the market.