Optimizing Besifloxacin Hydrochloride Production: A High-Yield Boron-Chelation Strategy for Commercial Scale
Optimizing Besifloxacin Hydrochloride Production: A High-Yield Boron-Chelation Strategy for Commercial Scale
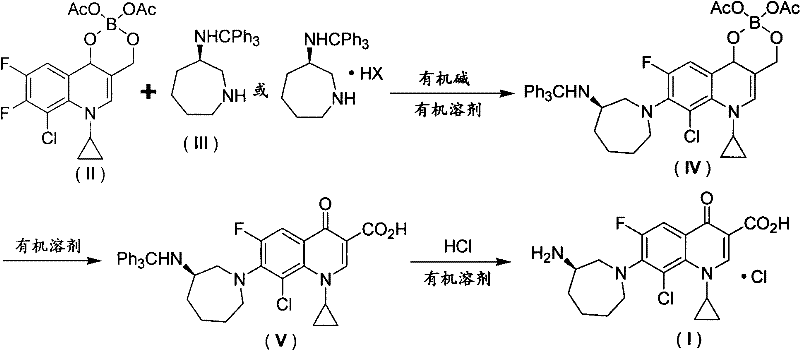
The pharmaceutical industry constantly seeks robust synthetic pathways that balance high purity with economic viability, particularly for complex fluoroquinolone antibiotics. Patent CN102659761A introduces a transformative methodology for the preparation of Besifloxacin Hydrochloride, a potent fourth-generation fluoroquinolone primarily used in ophthalmic formulations for treating bacterial conjunctivitis. This technical disclosure outlines a novel three-step sequence that leverages boron chelation chemistry to overcome the historical limitations of regioselectivity and yield associated with quinolone functionalization. By utilizing a 1-cyclopropyl-6,7-difluoro-8-chloro-quinoline-3-carboxylic acid-O3,O4-diacetate borate intermediate, the process effectively activates the 7-position for nucleophilic attack while shielding the sensitive 4-keto group. This strategic modification not only enhances the reaction efficiency but also simplifies the downstream purification processes, making it an ideal candidate for reliable pharmaceutical intermediate supplier networks aiming to secure stable API supply chains.

The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies for synthesizing Besifloxacin Hydrochloride have been plagued by significant inefficiencies that hinder large-scale commercial adoption. For instance, earlier approaches described in literature often relied on nitrobenzylidene protecting groups or Boc-protected amines, which presented substantial synthetic bottlenecks. The nitrobenzylidene route, while conceptually straightforward, frequently suffered from reproducibility issues and lacked detailed embodiment data, leading to inconsistent batch quality. More critically, the Boc-protection strategy reported by researchers like Wang Yajuan demonstrated alarmingly low yields, with condensation steps achieving only 58.2% efficiency and subsequent deprotection steps dropping to a mere 36.4%. Furthermore, the preparation of the requisite (R)-3-(tert-butoxycarbonylamino)-hexahydroazepine starting material was noted to be difficult and costly. These cumulative inefficiencies result in excessive waste generation, inflated production costs, and an inability to meet the stringent purity specifications required for ophthalmic-grade active ingredients, thereby creating a volatile supply environment for procurement managers.
The Novel Approach
In stark contrast, the methodology disclosed in CN102659761A employs a sophisticated boron-chelation strategy coupled with trityl protection to streamline the synthesis. The process initiates with the condensation of a boron-chelated quinolone derivative with (R)-3-tritylamino-hexahydroazepine or its stable salts. This approach fundamentally alters the electronic environment of the quinolone ring; the boron atom coordinates with the 4-keto and 3-carboxyl oxygens, withdrawing electron density and thereby activating the 7-fluorine atom for nucleophilic substitution. This activation allows the reaction to proceed under milder conditions with significantly improved selectivity, minimizing the formation of 6,7-disubstituted impurities. The use of the trityl group offers superior stability during the condensation phase compared to Boc groups, yet it remains easily removable under acidic conditions in the final step. This synergy between boron activation and robust amine protection results in a streamlined workflow that drastically reduces the number of purification cycles needed, directly addressing the cost reduction in pharmaceutical intermediates manufacturing by maximizing material throughput.
Mechanistic Insights into Boron-Chelated Nucleophilic Substitution
The core innovation of this synthesis lies in the mechanistic advantage provided by the O3,O4-diacetate borate moiety. In standard fluoroquinolone synthesis, the 4-keto group can compete for electrons or participate in side reactions, and the difference in reactivity between the 6-fluoro and 7-fluoro positions is often insufficient to guarantee exclusive 7-substitution. However, when the quinolone is converted into its boron chelate, the empty p-orbital of the boron atom accepts lone pair electrons from the carbonyl oxygen. This interaction creates a powerful electron-withdrawing effect that propagates through the aromatic system, specifically enhancing the electrophilicity of the carbon at the 7-position. Consequently, the nucleophilic attack by the hexahydroazepine nitrogen occurs with high precision. This electronic tuning not only accelerates the reaction kinetics, allowing completion within 2 to 8 hours at moderate temperatures (40°C to solvent boiling point), but also suppresses the formation of regioisomeric impurities. For R&D directors, this implies a much cleaner crude reaction profile, reducing the burden on analytical teams to identify and quantify trace impurities during method validation.
Furthermore, the choice of the trityl (triphenylmethyl) protecting group on the amine side chain is mechanistically sound for this specific application. Unlike smaller protecting groups that might sterically hinder the approach to the crowded 7-position of the quinolone, the trityl group, despite its bulk, is attached to the nitrogen in a way that allows the ring nitrogen of the hexahydroazepine to remain accessible for the initial condensation. More importantly, the trityl cation is highly stable, meaning the protecting group does not prematurely cleave under the basic conditions required for the nucleophilic substitution (using bases like triethylamine or DBU). It is only in the final stage, upon exposure to hydrochloric acid in a protic solvent, that the trityl group is cleaved to reveal the free amine, which immediately forms the hydrochloride salt. This orthogonal stability ensures that the intermediate remains intact throughout the harsh conditions of the coupling reaction, thereby preserving the stereochemical integrity of the (R)-enantiomer, which is critical for the biological activity of the final antibiotic.
How to Synthesize Besifloxacin Hydrochloride Efficiently
The synthesis protocol outlined in the patent provides a clear roadmap for transitioning from laboratory scale to commercial production. The process is divided into three distinct operational units: the condensation of the boron-chelated acid with the protected amine, the hydrolytic removal of the boron ester to regenerate the carboxylic acid, and the final acidic deprotection to yield the target salt. Each step utilizes common industrial solvents such as DMF, acetonitrile, and ethanol, avoiding exotic or highly hazardous reagents that would complicate safety compliance. The following guide summarizes the critical operational parameters derived from the patent examples, serving as a foundational reference for process engineers looking to implement this technology.
- Condense 1-cyclopropyl-6,7-difluoro-8-chloro-quinoline boron diacetate with (R)-3-tritylamino-hexahydroazepine using an organic base.
- Hydrolyze the resulting boron ester intermediate in an alcoholic solvent under reflux to obtain the carboxylic acid precursor.
- Treat the precursor with hydrochloric acid in an organic solvent to remove the trityl protecting group and form the final hydrochloride salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this boron-chelation route represents a strategic opportunity to stabilize the supply of high-value ophthalmic antibiotics. Traditional synthesis paths often suffer from yield attrition, where the cumulative loss across multiple low-efficiency steps drives up the cost of goods sold (COGS) and necessitates larger reactor volumes to meet demand. By implementing a route that demonstrates high conversion rates in each step—as evidenced by the patent's experimental data showing yields exceeding 75% for condensation and over 90% for subsequent steps—manufacturers can significantly optimize their production capacity. This efficiency translates directly into a more resilient supply chain, capable of absorbing market fluctuations without the need for excessive safety stock. Moreover, the use of stable, commercially available starting materials like trityl-protected amines reduces the risk of raw material shortages that often plague specialized synthetic routes.
- Cost Reduction in Manufacturing: The economic impact of this process is driven primarily by the elimination of low-yield steps and the reduction of solvent usage associated with extensive purification. In previous methods, the need to separate difficult impurities and the loss of material during the deprotection phase (often below 40% yield) created a massive financial drain. The new method's high-yield condensation and near-quantitative final deprotection mean that less raw material is required per kilogram of finished API. Additionally, the ability to use simpler work-up procedures, such as direct crystallization from ethanol rather than column chromatography, drastically lowers operational expenditures related to silica gel, solvents, and labor hours. This structural cost advantage allows suppliers to offer more competitive pricing models while maintaining healthy margins.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by the complexity of synthesizing key intermediates. The reliance on difficult-to-prepare Boc-protected amines in older routes introduced a single point of failure in the supply chain. By shifting to trityl-protected amines, which are more robust and easier to source or synthesize in bulk, the overall risk profile of the manufacturing process is lowered. The reaction conditions described, operating at moderate temperatures and atmospheric pressure, are compatible with standard glass-lined or stainless-steel reactors found in most multipurpose chemical plants. This compatibility ensures that the technology can be rapidly deployed across different manufacturing sites globally, mitigating the risk of regional production disruptions and ensuring a steady flow of high-purity pharmaceutical intermediates to downstream formulators.
- Scalability and Environmental Compliance: From an environmental and regulatory standpoint, this process offers distinct advantages regarding waste management and safety. The avoidance of nitro-containing intermediates eliminates the generation of potentially hazardous nitroso byproducts and simplifies wastewater treatment protocols. The solvents employed, such as ethanol and acetonitrile, are well-understood and easily recovered through distillation, supporting green chemistry initiatives and reducing the facility's environmental footprint. Furthermore, the high selectivity of the boron-chelated reaction minimizes the formation of genotoxic impurities or difficult-to-remove regioisomers, facilitating a smoother regulatory approval process. This ease of scale-up ensures that the transition from pilot plant batches to multi-ton commercial production can be achieved with minimal process re-engineering, securing long-term supply stability.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and claims within the patent documentation, providing clarity on the feasibility and advantages of the technology for potential partners and licensees.
Q: Why is the boron-chelation method superior to previous nitrobenzylidene or Boc-protection routes?
A: The boron-chelation method significantly improves regioselectivity at the 7-position of the quinolone ring, preventing unwanted substitution at the 6-position. Unlike previous methods which suffered from low yields (e.g., 36.4% for deprotection in Boc routes) and difficult starting material preparation, this route utilizes stable trityl protection and achieves high conversion rates suitable for industrial scaling.
Q: What are the critical reaction conditions for the condensation step?
A: The condensation requires an organic base such as triethylamine, pyridine, or DBU, and polar aprotic solvents like DMF or acetonitrile. The reaction temperature typically ranges from 40°C to the solvent's boiling point, ensuring complete conversion of the fluoroquinolone boron ester within 2 to 8 hours.
Q: How is the final purity and salt formation managed in this process?
A: The final step involves treating the trityl-protected precursor with hydrochloric acid in solvents like ethanol at mild temperatures (15-30°C). This simultaneously removes the robust trityl group and forms the stable hydrochloride salt, yielding a high-purity solid after simple filtration and washing, avoiding complex chromatographic purification.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Besifloxacin Hydrochloride Supplier
The technological advancements detailed in CN102659761A underscore the immense potential for optimizing the production of critical ophthalmic antibiotics. At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to translate such innovative patent methodologies into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of high-yield boron-chelation chemistry are fully realized in a GMP-compliant environment. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of Besifloxacin Hydrochloride meets the exacting standards required for global pharmaceutical markets.
We invite procurement leaders and R&D directors to collaborate with us to leverage this advanced synthesis route for their supply chains. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us directly to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that enhance both the quality and economic efficiency of your antibiotic portfolios.