Advanced Manufacturing of Tenofovir Disoproxil Fumarate Impurity T-C for Global Pharmaceutical Quality Control
Advanced Manufacturing of Tenofovir Disoproxil Fumarate Impurity T-C for Global Pharmaceutical Quality Control
The rigorous demand for high-purity reference standards in the pharmaceutical industry has driven significant innovation in the synthesis of complex nucleotide analogs. Patent CN110372750B introduces a robust and highly efficient methodology for the preparation of Tenofovir Disoproxil Fumarate Impurity T-C, a critical quality control marker for one of the world's most vital antiretroviral medications. This specific impurity, chemically defined as (R)-[[2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl]-(isopropoxy)-phosphonic acid isopropyloxycarbonyloxymethyl ester, serves as an essential benchmark for ensuring the safety and efficacy of Tenofovir Disoproxil Fumarate batches globally. By leveraging a novel two-step sequence involving precise esterification and condensation reactions, this technology addresses the historical challenges of low yield and difficult purification associated with earlier synthetic routes. The strategic implementation of EDC.HCl mediated coupling allows for exceptional control over the reaction profile, minimizing side products and maximizing the recovery of the target molecule. For R&D directors and quality assurance teams, access to such well-characterized impurities is not merely a regulatory checkbox but a fundamental component of comprehensive drug safety profiling.
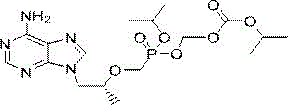
Understanding the molecular architecture of Impurity T-C is paramount for analytical chemists tasked with method validation. The structure features a chiral phosphonate center linked to an adenine base, protected by specific isopropoxycarbonyloxymethyl groups that mimic the prodrug functionality of the parent API. The presence of this specific impurity in final drug substances can indicate deviations in the manufacturing process, such as incomplete reactions or hydrolysis events. Therefore, the ability to synthesize this compound reliably and in high purity enables laboratories to establish accurate detection limits and quantification protocols. The patent highlights that this impurity is generated in most process routes for Tenofovir Disoproxil Fumarate, making its availability as a certified reference material crucial for regulatory submissions in major markets including the US, Europe, and China. As regulatory agencies tighten their scrutiny on impurity profiles, having a dependable source for such specific degradation products becomes a strategic asset for any pharmaceutical manufacturer aiming for market approval.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of phosphorylated nucleotide impurities has been plagued by harsh reaction conditions and cumbersome purification workflows that hinder scalability. Prior art methods, such as those disclosed in earlier patents like CN 104341452A, often relied on reaction pathways that resulted in suboptimal yields and required extensive chromatographic separation to achieve acceptable purity levels. These traditional approaches frequently utilized coupling agents that generated insoluble byproducts, such as dicyclohexylurea (DCU), which are notoriously difficult to remove completely from the reaction mixture without significant product loss. Furthermore, the use of aggressive reagents or extreme temperatures in older protocols could lead to racemization at the chiral center or degradation of the sensitive purine ring system. Such inefficiencies not only drive up the cost of goods but also introduce variability that is unacceptable for the production of reference standards. The reliance on multiple recrystallization steps or preparative HPLC significantly extends the production timeline, creating bottlenecks in the supply chain for critical quality control materials.
The Novel Approach
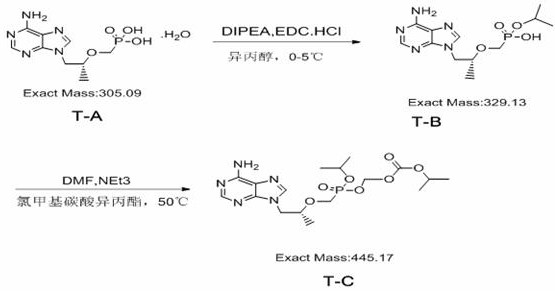
The methodology outlined in patent CN110372750B represents a paradigm shift towards greener and more efficient chemical manufacturing for this specific class of impurities. By utilizing T-A as a readily available starting material and employing EDC.HCl in conjunction with DIPEA, the process achieves a highly selective esterification under mild conditions ranging from 0-5°C. This低温 strategy effectively suppresses thermal degradation and preserves the stereochemical integrity of the molecule. The subsequent condensation step utilizes chloromethyl isopropyl carbonate in a polar aprotic solvent, facilitating the formation of the carbonate ester linkage with high conversion rates. A key innovation lies in the workup procedure, where pH manipulation is used to induce precipitation of the intermediate T-B, thereby eliminating the need for column chromatography at this stage. This streamlined approach not only enhances the overall throughput but also significantly reduces the consumption of organic solvents and silica gel, aligning with modern principles of sustainable chemistry.
Mechanistic Insights into EDC-Mediated Esterification and Condensation
The core of this synthetic success lies in the mechanistic efficiency of the carbodiimide-mediated activation of the phosphonic acid moiety. In the first step, EDC.HCl acts as a dehydrating agent, reacting with the phosphonic acid group of T-A to form a highly reactive O-acylisourea intermediate. This activated species is then susceptible to nucleophilic attack by the alcohol oxygen, leading to the formation of the phosphonate monoester T-B. The presence of DIPEA serves a dual purpose: it acts as a proton scavenger to neutralize the hydrochloric acid released during the activation and maintains the basicity required to drive the equilibrium forward. Unlike DCC, the urea byproduct formed from EDC is water-soluble, which is a critical design feature that simplifies the downstream processing. The reaction is conducted in isopropanol, which serves both as a solvent and potentially participates in the solvation shell, stabilizing the transition states. The careful control of temperature at 0-5°C ensures that the activation energy is sufficient for the desired transformation while minimizing competing hydrolysis of the activated intermediate.
In the second stage, the conversion of T-B to the final Impurity T-C involves a nucleophilic substitution reaction where the remaining hydroxyl group attacks the chloromethyl isopropyl carbonate. The use of triethylamine or DIPEA as an acid-binding agent is essential here to trap the hydrogen chloride generated during the displacement of the chloride ion. The reaction proceeds smoothly in DMF at moderate temperatures of 45-50°C, a range that provides adequate kinetic energy for the reaction without compromising the stability of the thermally sensitive purine base. The mechanism likely proceeds via an SN2 pathway, where the alkoxide anion, generated in situ by the base, displaces the chloride leaving group. The high purity of the final product (>98%) suggests that the reaction is highly chemoselective, with minimal formation of bis-alkylated byproducts or hydrolysis of the newly formed carbonate ester. This selectivity is crucial for avoiding complex mixtures that would require expensive and time-consuming purification techniques.
How to Synthesize Tenofovir Disoproxil Fumarate Impurity T-C Efficiently
The synthesis protocol described in the patent offers a reproducible framework for producing high-quality Impurity T-C suitable for analytical applications. The process begins with the dissolution of the starting material T-A in isopropanol, followed by the sequential addition of the coupling reagents under an inert nitrogen atmosphere to prevent moisture interference. The reaction mixture is stirred for an extended period of 12 hours to ensure complete conversion, after which the solvent is removed to yield an oily concentrate. The workup involves a biphasic extraction system using dichloromethane and water, where pH adjustments play a pivotal role in isolating the intermediate T-B as a solid precipitate. This solid is then carried forward directly into the second step without further purification, demonstrating the robustness of the method. The final condensation is monitored by TLC to determine the endpoint, ensuring that the reaction is stopped before any degradation occurs. For a detailed, step-by-step breakdown of the exact molar ratios, solvent volumes, and specific handling instructions, please refer to the standardized guide below.
- Perform esterification of T-A using EDC.HCl and DIPEA in isopropanol at 0-5°C to obtain intermediate T-B.
- Purify T-B via pH adjustment and crystallization, achieving over 92% purity without chromatography.
- Conduct condensation of T-B with chloromethyl isopropyl carbonate in DMF at 45-50°C to yield final Impurity T-C.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this synthesis route offers tangible benefits that extend beyond mere technical feasibility. The reliance on commodity chemicals such as isopropanol, DMF, and triethylamine ensures that the raw material supply is stable and not subject to the volatility often seen with exotic reagents. This accessibility translates directly into reduced lead times for production planning and a lower risk of supply disruptions. Furthermore, the elimination of chromatographic purification for the intermediate step significantly lowers the operational expenditure associated with silica gel and large volumes of elution solvents. The ability to isolate the intermediate via simple pH adjustment and filtration is a major cost-saving driver, as it reduces both the labor hours required for processing and the waste disposal costs associated with solvent-heavy purification methods. For supply chain managers, this means a more predictable and cost-effective manufacturing cycle that can be scaled up to meet fluctuating market demands without proportional increases in overhead.
- Cost Reduction in Manufacturing: The process achieves significant economic efficiency by replacing expensive and difficult-to-remove coupling byproducts with water-soluble alternatives. By utilizing EDC.HCl instead of DCC, the need for extensive filtration and washing steps to remove insoluble ureas is completely eliminated, resulting in substantial savings in labor and solvent usage. Additionally, the high yield of the reaction minimizes the amount of starting material wasted, directly improving the cost per gram of the final impurity standard. The recycling of solvents like ethyl acetate in the final workup further contributes to a leaner manufacturing budget, allowing for competitive pricing strategies in the global market for reference materials.
- Enhanced Supply Chain Reliability: The use of widely available starting materials like T-A and common organic solvents mitigates the risk of supply chain bottlenecks. Unlike processes that depend on specialized catalysts or custom-synthesized reagents, this method relies on a supply chain that is robust and well-established globally. The mild reaction conditions also reduce the wear and tear on reactor equipment, lowering maintenance costs and extending the lifespan of manufacturing assets. This reliability ensures that production schedules can be met consistently, providing customers with a steady stream of high-quality impurity standards necessary for their own regulatory compliance and batch release testing.
- Scalability and Environmental Compliance: The synthetic route is inherently scalable, moving seamlessly from gram-scale laboratory synthesis to kilogram-scale commercial production without the need for re-optimization. The reduction in solvent usage and the avoidance of hazardous heavy metal catalysts align with increasingly stringent environmental regulations regarding waste discharge. The simplified workup procedures generate less wastewater and organic waste, reducing the environmental footprint of the manufacturing process. This commitment to green chemistry not only fulfills corporate social responsibility goals but also future-proofs the supply chain against tightening environmental legislation, ensuring long-term operational continuity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of Tenofovir Disoproxil Fumarate Impurity T-C. These answers are derived directly from the experimental data and claims presented in patent CN110372750B, providing a reliable basis for decision-making. Understanding these details helps stakeholders assess the suitability of this material for their specific analytical needs and quality control frameworks. For further technical discussions or custom synthesis requests, our team is available to provide deeper insights.
Q: What is the purity level achievable with this synthesis method?
A: The patented process consistently delivers Impurity T-C with an HPLC purity greater than 98%, meeting stringent requirements for reference standards.
Q: Why is EDC.HCl preferred over DCC in this reaction?
A: EDC.HCl generates water-soluble urea byproducts that are easily removed during aqueous workup, unlike the insoluble DCU formed by DCC, simplifying purification.
Q: Is this process scalable for commercial production?
A: Yes, the mild reaction conditions (0-50°C) and use of common solvents like isopropanol and DMF make the process highly adaptable for large-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tenofovir Disoproxil Fumarate Impurity T-C Supplier
At NINGBO INNO PHARMCHEM, we understand that the integrity of your pharmaceutical products depends on the quality of the reference standards you use. Our expertise in fine chemical synthesis allows us to replicate and optimize complex pathways like the one described in CN110372750B, ensuring that you receive materials of the highest purity and consistency. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, backed by state-of-the-art facilities and rigorous QC labs that adhere to international standards. Our commitment to excellence means that every batch of Tenofovir Disoproxil Fumarate Impurity T-C is thoroughly characterized using advanced analytical techniques, guaranteeing that it meets the stringent specifications required by global regulatory bodies.
We invite you to collaborate with us to secure a stable and cost-effective supply of this critical impurity. By leveraging our technical capabilities, you can streamline your quality control processes and accelerate your drug development timelines. Please contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our solutions can add value to your supply chain and support your commitment to patient safety.