Strategic Synthesis and Commercial Scale-Up of 8-Halogenated Adenine Antiviral Intermediates
Strategic Synthesis and Commercial Scale-Up of 8-Halogenated Adenine Antiviral Intermediates
The pharmaceutical landscape for antiviral therapeutics is constantly evolving, driven by the need for agents with superior bioavailability and reduced resistance profiles. Patent CN101003550B introduces a significant advancement in this domain by disclosing a series of 8-halogenated adenine acyclic nucleoside compounds and their organic acid salts. These molecules, structurally related to established antivirals like Tenofovir and Adefovir, represent a strategic evolution in nucleotide analog design. By modifying the purine base at the 8-position with halogens such as fluorine, chlorine, or bromine, the invention addresses the critical limitation of poor lipophilicity inherent in many phosphate prodrugs. This technical disclosure provides a robust foundation for developing next-generation treatments for HIV and HBV infections, offering a compelling value proposition for pharmaceutical developers seeking reliable antiviral pharmaceutical intermediate suppliers.
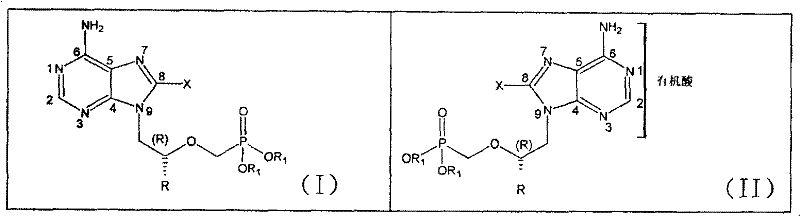
The core innovation lies in the specific structural configuration where the halogen atom is positioned at the C8 location of the adenine ring, while the phosphonate side chain remains intact or modified with lipophilic esters. The patent details the preparation of these compounds through a direct halogenation strategy, followed by salt formation with organic acids like fumaric acid. This approach not only stabilizes the active pharmaceutical ingredient but also optimizes its physicochemical properties for oral administration. For R&D directors evaluating new chemical entities, this patent offers a clear pathway to enhance the therapeutic index of existing nucleoside scaffolds without necessitating a complete de novo drug discovery program.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional nucleoside reverse transcriptase inhibitors, such as the parent compounds Adefovir and Tenofovir, often suffer from suboptimal pharmacokinetic properties due to their high polarity and strong hydrophilicity. While prodrug strategies like dipivoxil esters have been employed to improve membrane permeability, there remains a ceiling to how much bioavailability can be enhanced solely through side-chain modification. Conventional synthesis methods for nucleoside analogs frequently involve complex protection and deprotection sequences, leading to lower overall yields and higher production costs. Furthermore, the lack of structural diversity in the purine base limits the ability to fine-tune the electronic properties of the molecule, which can be crucial for overcoming viral resistance mechanisms. These factors collectively create a bottleneck in the cost reduction in nucleoside analog manufacturing, prompting the industry to seek more efficient synthetic alternatives.
The Novel Approach
The methodology outlined in CN101003550B circumvents these challenges by introducing a halogen substituent directly onto the adenine ring, a modification that fundamentally alters the electron density and lipophilic character of the molecule. This novel approach utilizes a straightforward electrophilic substitution reaction in acetic acid, eliminating the need for harsh conditions or exotic reagents. The subsequent formation of organic acid salts, particularly fumarates, further refines the solid-state properties of the drug substance, facilitating better formulation stability and dissolution rates. By focusing on the 8-position, the synthesis avoids interference with the N9-glycosidic bond or the phosphonate moiety, ensuring high regioselectivity. This streamlined process represents a significant leap forward in the commercial scale-up of complex antiviral intermediates, offering a more economically viable route to high-purity active ingredients.
Mechanistic Insights into Electrophilic Halogenation of Adenine Derivatives
The chemical transformation at the heart of this technology is an electrophilic aromatic substitution on the imidazole portion of the purine system. In the presence of a mild base such as sodium acetate, the adenine ring becomes activated towards electrophilic attack. When a halogen source like molecular bromine (Br2) or chlorine (Cl2) is introduced, the electron-rich C8 position acts as the nucleophile, attacking the halogen molecule to form a sigma complex. The acetate ion then facilitates the restoration of aromaticity by abstracting the proton from the C8 position, resulting in the stable 8-halo product. This mechanism is highly efficient and proceeds readily at temperatures ranging from 0°C to 100°C, with room temperature often being sufficient for bromination. The choice of solvent, typically acetic acid or alcohols like methanol, plays a dual role in solubilizing the polar nucleoside precursor and stabilizing the transition state of the reaction.
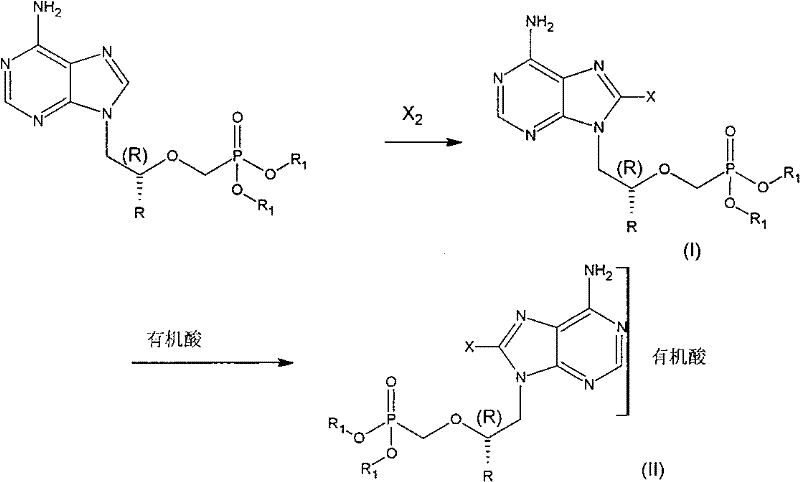
Impurity control is inherently managed by the specificity of the electrophilic substitution. Since the C8 position is the most reactive site on the adenine ring for electrophilic attack, poly-halogenation or substitution at other positions is minimized. The workup procedure described in the patent involves quenching excess halogen with sodium bisulfite, which prevents oxidative degradation of the product during isolation. Adjusting the aqueous phase pH to 8.0-8.5 ensures that the basic nitrogen atoms of the purine ring are deprotonated, maximizing the extraction efficiency into organic solvents like ethyl acetate. This precise control over reaction parameters and workup conditions ensures that the final 8-halogenated intermediate meets stringent purity specifications required for pharmaceutical applications, reducing the burden on downstream purification processes.
How to Synthesize 8-Halogenated Adenine Nucleoside Efficiently
The synthesis protocol described in the patent provides a reproducible framework for producing these valuable intermediates. The process begins with the dissolution of the adenine acyclic nucleoside precursor in a suitable solvent system, followed by the controlled addition of the halogenating agent. Reaction progress is monitored via TLC to ensure complete conversion of the starting material. Once the reaction is complete, a systematic workup involving extraction, washing, and drying yields the crude 8-halo compound.
- Dissolve the adenine acyclic nucleoside precursor in acetic acid or alcohol, add a base like sodium acetate, and introduce halogen source (Br2, Cl2, or F2/He) at controlled temperatures.
- Quench the reaction with sodium bisulfite, extract the organic phase, adjust pH to 8.0-8.5, and isolate the 8-halogenated intermediate via solvent evaporation.
- React the isolated intermediate with an organic acid such as fumaric acid in methanol or ethanol under reflux to crystallize the final stable salt form.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the synthetic route detailed in this patent offers substantial advantages over traditional multi-step nucleoside syntheses. The reliance on commodity chemicals such as acetic acid, sodium acetate, and elemental halogens means that raw material sourcing is straightforward and less susceptible to geopolitical supply disruptions. The elimination of expensive transition metal catalysts, which often require specialized removal steps to meet residual metal limits, translates directly into simplified processing and reduced waste management costs. This simplicity allows for a more agile supply chain capable of responding quickly to market demands for antiviral therapies.
- Cost Reduction in Manufacturing: The process achieves cost efficiency by utilizing inexpensive reagents and avoiding complex catalytic systems. The direct halogenation step replaces multiple protection-deprotection cycles found in older methodologies, significantly shortening the production timeline. Furthermore, the high regioselectivity of the reaction minimizes the formation of difficult-to-separate isomers, thereby improving overall yield and reducing the volume of solvents required for purification. These factors combine to lower the cost of goods sold (COGS) for the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The robustness of the chemistry ensures consistent batch-to-batch quality, which is critical for maintaining regulatory compliance. Since the reaction conditions are mild and do not require extreme pressures or cryogenic temperatures, the risk of equipment failure or process deviation is minimized. This reliability fosters a stable supply of high-purity 8-halogenated adenine derivatives, enabling pharmaceutical partners to plan their production schedules with greater confidence and reducing lead time for high-purity antiviral intermediates.
- Scalability and Environmental Compliance: The synthesis is inherently scalable, as demonstrated by the use of standard unit operations like liquid-liquid extraction and crystallization. The waste streams generated are primarily aqueous salts and organic solvents, which can be treated using conventional wastewater management systems. The absence of heavy metals simplifies environmental compliance and reduces the ecological footprint of the manufacturing process. This aligns with modern green chemistry principles, making the technology attractive for companies aiming to enhance their sustainability profiles while expanding production capacity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these 8-halogenated compounds. The answers are derived directly from the experimental data and claims presented in the patent documentation, ensuring accuracy and relevance for technical decision-makers.
Q: How does 8-halogenation improve the pharmacokinetic profile of tenofovir derivatives?
A: The introduction of a halogen atom at the 8-position of the adenine ring significantly increases the lipophilicity of the molecule. This structural modification enhances intestinal epithelial penetration and oral bioavailability compared to the parent non-halogenated compounds, addressing the poor absorption issues often seen with highly hydrophilic nucleotide analogs.
Q: What are the critical safety considerations for the halogenation step in this process?
A: The process utilizes elemental halogens such as bromine or chlorine, which require strict handling protocols. The reaction is typically conducted in acetic acid with a base like sodium acetate to moderate reactivity. Proper quenching with sodium bisulfite is essential to neutralize excess halogen before workup, ensuring operator safety and preventing downstream contamination.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the route is designed for scalability. It relies on commodity chemicals like acetic acid, sodium acetate, and elemental bromine, avoiding expensive transition metal catalysts. The workup involves standard liquid-liquid extraction and crystallization, which are unit operations easily transferable from laboratory to multi-ton industrial reactors without complex purification challenges.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 8-Halogenated Adenine Nucleoside Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of life-saving antiviral medications. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising chemistry described in CN101003550B can be translated into reality. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 8-halogenated adenine nucleoside meets the highest international standards. Our commitment to excellence extends beyond mere synthesis; we provide comprehensive support to navigate the complexities of regulatory filings and process validation.
We invite pharmaceutical partners to collaborate with us to unlock the full potential of this technology. By leveraging our expertise in process optimization, we can help you achieve a Customized Cost-Saving Analysis tailored to your specific production volumes. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments. Together, we can accelerate the delivery of improved antiviral therapies to patients worldwide, combining scientific innovation with operational excellence.