Advanced Catalytic Route for Scalable 6-Difluorophenanthridine Pharmaceutical Intermediates Production
Advanced Catalytic Route for Scalable 6-Difluorophenanthridine Pharmaceutical Intermediates Production
The landscape of fluorinated heterocycle synthesis has been significantly advanced by the disclosures in patent CN108675960B, which details a robust catalytic method for producing 6-difluorophenanthridine compounds. These fluorinated scaffolds are increasingly critical in modern medicinal chemistry due to their unique metabolic stability and lipophilicity profiles, making them highly sought-after building blocks for next-generation active pharmaceutical ingredients (APIs). The patented technology addresses a longstanding challenge in organic synthesis: the efficient activation and transformation of stable carbon-fluorine bonds to construct complex polycyclic systems. By leveraging a copper-catalyzed defluorinative cyclization strategy, this process transforms simple, commercially available o-aminobiphenyl derivatives and ethyl bromodifluoroacetate into valuable phenanthridine cores with high efficiency. For R&D directors and procurement specialists in the fine chemical sector, this represents a pivotal shift towards more sustainable and cost-effective manufacturing pathways for high-value fluorinated intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of difluoromethylated heterocycles has relied on multi-step sequences involving harsh reagents or pre-functionalized starting materials that are often expensive and hazardous to handle. Traditional difluoroalkylation strategies frequently struggle with the thermodynamic stability of C-F bonds, requiring extreme conditions that compromise functional group tolerance and lead to complex impurity profiles. Furthermore, many existing methods necessitate the use of specialized fluorine sources that are not readily available on a industrial scale, creating bottlenecks in the supply chain for pharmaceutical intermediates. The inability to efficiently cleave multiple bonds on a single carbon center in a concerted manner has limited the structural diversity accessible to chemists, often forcing reliance on linear syntheses that suffer from poor atom economy and low overall yields. These limitations translate directly into higher production costs and extended lead times for drug development projects requiring fluorinated phenanthridine motifs.
The Novel Approach
In stark contrast, the methodology described in CN108675960B introduces a streamlined, one-pot catalytic cycle that elegantly overcomes these barriers by utilizing ethyl bromodifluoroacetate as a versatile C1 source. This novel approach facilitates the simultaneous cleavage of C-Br and C-COOM bonds alongside the activation of C-F bonds, enabling the direct formation of the phenanthridine skeleton under relatively mild thermal conditions. The reaction system employs a copper(II) triflate catalyst paired with a 1,10-phenanthroline ligand, which synergistically promotes the defluorinative cyclization with remarkable selectivity. As illustrated in the general reaction scheme below, the process converts o-aminobiphenyl substrates directly into the target 6-difluorophenanthridine esters, bypassing the need for isolated intermediates.
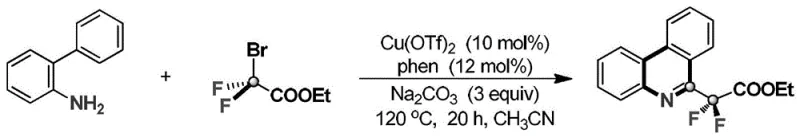
The operational simplicity of this new route is a major advantage, as it utilizes standard laboratory equipment and common solvents like acetonitrile, eliminating the need for specialized high-pressure reactors or cryogenic setups often associated with fluorine chemistry. This accessibility drastically reduces the barrier to entry for scaling the process from gram to kilogram quantities, offering a compelling alternative for manufacturers seeking to optimize their production of fluorinated heterocycles.
Mechanistic Insights into Cu-Catalyzed Defluorinative Cyclization
From a mechanistic perspective, the success of this transformation lies in the unique ability of the copper catalyst to mediate the activation of the inert C-F bonds adjacent to the ester functionality. The proposed catalytic cycle likely involves the initial oxidative addition or single-electron transfer to the carbon-bromine bond of the ethyl bromodifluoroacetate, generating a reactive difluorocarbene or radical species in situ. This reactive intermediate then engages with the ortho-amino group of the biphenyl substrate, initiating a cascade that ultimately results in the formation of the new C-C and C-N bonds required to close the phenanthridine ring. The presence of the phenanthroline ligand is crucial, as it stabilizes the copper center in the appropriate oxidation state and prevents catalyst deactivation, ensuring high turnover numbers throughout the reaction duration of 10 to 25 hours at 120 °C.
Furthermore, the reaction exhibits exceptional functional group compatibility, a critical factor for R&D teams designing diverse libraries of drug candidates. The protocol tolerates a wide array of substituents on the biphenyl backbone, including electron-donating groups like methoxy and alkyl chains, as well as electron-withdrawing groups such as halogens and esters. The extensive substrate scope demonstrated in the patent data highlights the robustness of this chemistry, with yields consistently ranging from 63% to 88% across various derivatives. This broad tolerance minimizes the need for protecting group strategies, thereby shortening synthetic routes and reducing waste generation. The ability to access such a diverse range of fluorinated scaffolds from a single set of reaction conditions underscores the versatility of this platform for generating high-purity pharmaceutical intermediates.
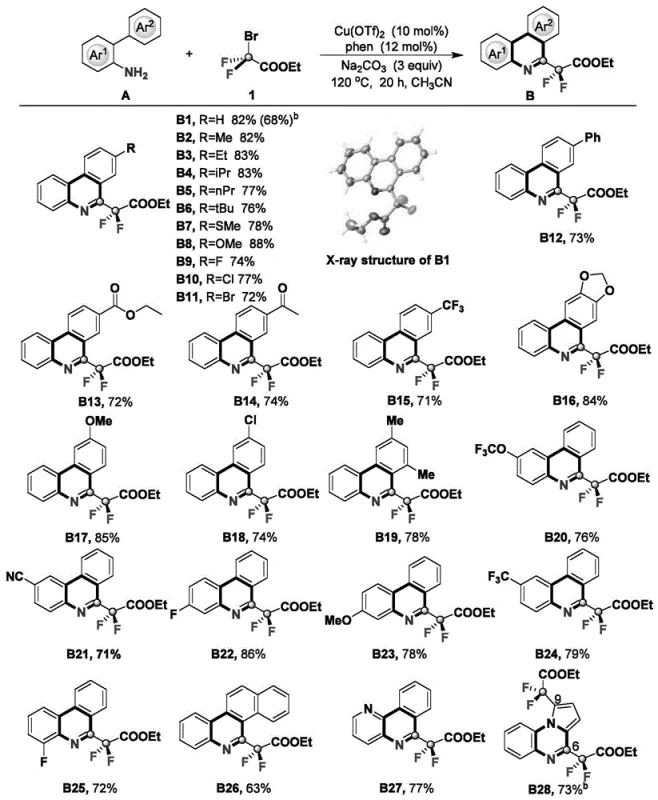
How to Synthesize 6-Difluorophenanthridine Efficiently
The synthesis of these valuable fluorinated intermediates is designed for operational ease, requiring only standard mixing and heating capabilities found in most pilot and production plants. The process begins with the precise charging of o-aminobiphenyl, ethyl bromodifluoroacetate, the copper catalyst, ligand, and base into a pressure-resistant vessel under an inert nitrogen atmosphere. Following the reaction period, the workup involves a straightforward filtration and extraction sequence, avoiding complex quenching procedures that often generate hazardous waste streams. For detailed operational parameters and safety guidelines, please refer to the standardized synthesis steps outlined below.
- Combine o-aminobiphenyl compound, ethyl bromodifluoroacetate, Cu(OTf)2, 1,10-phenanthroline, sodium carbonate, and acetonitrile in a sealed vessel under nitrogen.
- Heat the mixture in an oil bath at 120 °C for 10 to 25 hours while monitoring reaction progress via TLC or GC.
- Cool to room temperature, filter, extract with ethyl acetate, concentrate, and purify via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this catalytic technology offers substantial strategic benefits beyond mere chemical efficiency. The reliance on commodity chemicals such as sodium carbonate, acetonitrile, and commercially sourced ethyl bromodifluoroacetate ensures a stable and resilient supply chain, mitigating the risks associated with sourcing exotic or proprietary reagents. The elimination of multi-step sequences and protecting group manipulations translates directly into reduced raw material consumption and lower waste disposal costs, contributing to a significantly greener manufacturing profile. Moreover, the high selectivity of the reaction minimizes the formation of difficult-to-remove impurities, which simplifies downstream purification and enhances the overall throughput of the production facility.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the use of inexpensive, bulk-available starting materials and a catalyst system that operates at low loading levels (10 mol%). By consolidating what would traditionally be a multi-step synthesis into a single pot operation, manufacturers can realize drastic reductions in labor, energy, and solvent usage. The avoidance of cryogenic conditions or high-pressure hydrogenation further lowers capital expenditure requirements for equipment, allowing for cost-effective production even in facilities with standard infrastructure. These cumulative efficiencies result in a markedly lower cost of goods sold (COGS) for the final fluorinated intermediate.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions ensures consistent batch-to-batch reproducibility, a critical metric for maintaining uninterrupted supply to downstream API manufacturers. Since the reagents are not subject to the same supply volatility as specialized fluorinating agents, procurement teams can secure long-term contracts with greater confidence. The simplified logistics of handling non-hazardous solids and common solvents also streamline warehousing and transportation, reducing the administrative burden and regulatory compliance costs associated with dangerous goods.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated to proceed effectively in sealed vessels that mimic industrial autoclave conditions. The use of acetonitrile, a solvent with well-established recovery and recycling protocols, aligns with modern environmental, health, and safety (EHS) standards. Additionally, the high atom economy of the defluorinative cyclization means less chemical waste is generated per kilogram of product, facilitating easier compliance with increasingly stringent environmental regulations regarding fluorine-containing effluents.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this catalytic synthesis route. These insights are derived directly from the experimental data and beneficial effects reported in the patent literature, providing a factual basis for evaluating the technology's fit within your existing manufacturing portfolio. Understanding these nuances is essential for making informed decisions about process adoption and supplier qualification.
Q: What are the key advantages of this Cu-catalyzed method over traditional difluoroalkylation?
A: This method uniquely achieves the cleavage of four bonds on a single carbon atom in one step, utilizing readily available ethyl bromodifluoroacetate instead of complex fluorine sources, significantly simplifying the synthetic route.
Q: Is this process suitable for large-scale manufacturing of pharmaceutical intermediates?
A: Yes, the process uses common solvents like acetonitrile and stable catalysts, operates at moderate temperatures (120 °C), and involves simple workup procedures, making it highly amenable to commercial scale-up.
Q: What is the functional group tolerance of this synthesis?
A: The reaction demonstrates excellent functional group compatibility, tolerating various substituents such as halogens, esters, ethers, and alkyl groups on the biphenyl scaffold without significant yield loss.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-Difluorophenanthridine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the catalytic synthesis method described in CN108675960B for the production of high-value fluorinated heterocycles. As a leading CDMO partner, we possess the technical expertise and infrastructure to translate this laboratory-scale innovation into commercial reality, ensuring that our clients receive a consistent supply of high-purity intermediates. Our facilities are equipped to handle complex catalytic processes with rigorous quality control measures, guaranteeing that every batch meets the stringent purity specifications required for pharmaceutical applications. We have extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, allowing us to support your project from early-stage development through to full-scale manufacturing.
We invite you to collaborate with us to leverage this advanced chemistry for your specific drug development programs. Our technical team is ready to perform a Customized Cost-Saving Analysis tailored to your target molecule, demonstrating exactly how this route can optimize your budget and timeline. Please contact our technical procurement team today to request specific COA data for our reference standards and to discuss route feasibility assessments for your custom synthesis needs. Let us help you accelerate your pipeline with reliable, cost-effective, and scalable chemical solutions.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →