Scaling Low-Cardiotoxic Anthracycline Intermediates: A Technical Breakdown of Patent CN1154652C
Scaling Low-Cardiotoxic Anthracycline Intermediates: A Technical Breakdown of Patent CN1154652C
The development of next-generation oncology therapeutics often hinges on the ability to modify established scaffolds to mitigate severe side effects while retaining potency. Patent CN1154652C presents a pivotal advancement in the synthesis of 5-imino-13-deoxy anthracycline derivatives, a class of compounds designed specifically to overcome the dose-limiting cardiotoxicity associated with traditional agents like doxorubicin and daunorubicin. For R&D directors and process chemists, understanding the nuanced structural modifications detailed in this patent is crucial for developing safer API candidates. The core innovation lies in the dual modification of the anthracycline aglycone: the removal of the 13-keto group to prevent metabolic reduction to toxic 13-dihydro metabolites, and the conversion of the 5-keto group to an imino function to suppress free radical generation. This report provides a deep technical and commercial analysis of this synthesis route, positioning it as a viable strategy for reliable pharmaceutical intermediate supplier partnerships aiming to deliver high-purity oncology building blocks.
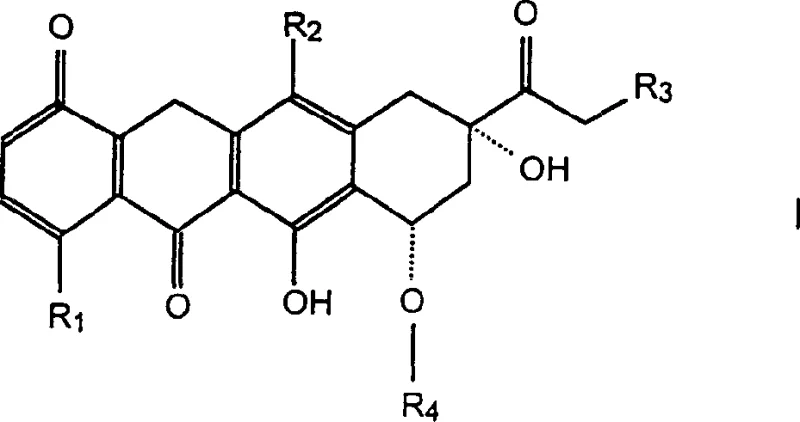
The structural versatility shown in the general formula allows for significant optimization of pharmacokinetic properties. By manipulating the R1, R2, and R3 substituents, as well as the glycosidic linkage at R4, medicinal chemists can fine-tune the solubility and cellular uptake of these potent antineoplastic agents. The patent explicitly notes that compounds containing morpholino-substituted sugar groups exhibit significantly increased toxicity, highlighting the importance of precise sugar moiety selection during the design phase. This level of structural control is essential for creating a robust impurity profile and ensuring that the final API meets stringent regulatory standards for clinical use.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the clinical utility of anthracycline antibiotics has been severely constrained by their narrow therapeutic index, primarily driven by cumulative, dose-dependent cardiotoxicity. Conventional synthesis methods focus on the native structures of doxorubicin and daunorubicin, which possess a 13-keto group on the D-ring. Extensive pharmacological studies cited in the background of the patent reveal that this specific ketone functionality is metabolically reduced in vivo to 13-dihydro metabolites, such as doxorubicinol. These metabolites are not merely inactive byproducts; they are potent cardiotoxins that cause persistent cardiac dysfunction and congestive heart failure even at therapeutic doses. Furthermore, the benzoquinone portion of the traditional molecule generates free radicals, contributing to oxidative stress in myocardial tissue. Consequently, the total lifetime dose for patients is capped at approximately 550 mg/m², limiting the efficacy of treatment in resistant cancers. From a manufacturing perspective, synthesizing analogs that retain potency without these liabilities requires breaking away from the standard 13-keto-5-keto paradigm, a challenge that conventional alkylation or acylation strategies have struggled to address efficiently.
The Novel Approach
The methodology outlined in CN1154652C offers a transformative solution by fundamentally altering the metabolic fate of the molecule through strategic deoxygenation and amination. Instead of attempting to shield the 13-keto group, the novel approach eliminates it entirely, creating a 13-deoxy scaffold that cannot be metabolized into the toxic 13-dihydro form. Simultaneously, the 5-keto group is converted into a 5-imino function, which significantly alters the redox potential of the quinone system, thereby reducing the propensity for free radical formation. This dual-modification strategy effectively decouples antitumor activity from cardiotoxicity. The synthetic route utilizes a tosylhydrazone intermediate, which serves as a versatile precursor for the reductive removal of the oxygen atom at the C-13 position. This approach not only solves the biological safety issue but also opens new avenues for cost reduction in API manufacturing by utilizing robust, scalable reduction chemistry that avoids the need for exotic catalysts or extreme cryogenic conditions often required for selective functionalization of complex polyketides.
Mechanistic Insights into Reductive Deoxygenation and Imino Formation
The core chemical transformation in this patent involves the reductive cleavage of the 13-tosylhydrazone derivative to yield the 13-deoxy anthracycline. This reaction is mechanistically distinct from standard hydride reductions due to the specific requirement for acidic conditions. The process begins with the formation of the tosylhydrazone from the parent 13-keto anthracycline. Crucially, the subsequent reduction with sodium cyanoborohydride must be conducted in an acidic medium, typically using p-toluenesulfonic acid in anhydrous methanol. Under these conditions, the tosylhydrazone is protonated, facilitating the elimination of nitrogen and the tosyl group to generate a reactive carbocation or diazenium intermediate at the C-13 position, which is then trapped by the hydride source. Maintaining the pH at 7.0 or lower is critical; basic conditions, often used in other hydrazone reductions, lead to rapid decomposition of the sensitive anthracycline chromophore. The reaction temperature is tightly controlled between 68°C and 72°C. This thermal window is narrow; temperatures below this range result in incomplete conversion, while temperatures exceeding 72°C induce thermal degradation of the tetracyclic core. This precise control ensures high selectivity and minimizes the formation of over-reduced byproducts or epimers at the C-13 stereocenter.
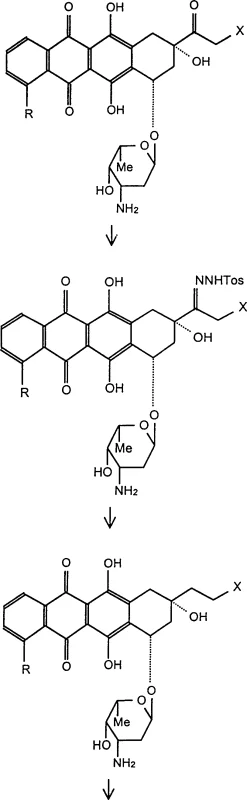
Following the successful deoxygenation, the synthesis proceeds to the installation of the 5-imino group, a step that requires careful protection group strategy to maintain chemoselectivity. The purified 13-deoxy intermediate is first protected at the 3'-amino position of the sugar moiety using a tert-butoxycarbonyl (Boc) group. This protection is vital to prevent unwanted side reactions during the subsequent ammoniation step. The 5-keto group is then reacted with ammonia under anhydrous conditions at low temperatures (0-4°C) to form the imine. The low temperature is essential to stabilize the imine bond and prevent hydrolysis or polymerization. Finally, the Boc group is removed under mild acidic conditions to reveal the free amine on the sugar, yielding the final 5-imino-13-deoxy anthracycline hydrochloride. This sequence demonstrates a high level of orthogonality in protecting group manipulation, ensuring that the final product possesses the desired substitution pattern without compromising the integrity of the glycosidic bond or the aglycone backbone.
How to Synthesize 5-Imino-13-Deoxydoxorubicin Efficiently
The practical execution of this synthesis requires strict adherence to anhydrous techniques and precise thermal management to achieve the reported yields of 50%-60% overall. The process begins with the preparation of the tosylhydrazone, followed by the critical acidic reduction step where the reaction mixture is gently refluxed. Post-reaction workup involves a unique quenching procedure where water is added to the cooled mixture to hydrolyze excess reagents, followed by extraction with halocarbon solvents like chloroform. The crude product is then subjected to rigorous purification, initially via silica gel chromatography to remove hydrophobic impurities, and subsequently via preparative HPLC using phenyl columns to separate the target compound from closely related analogs. The detailed operational parameters, including solvent ratios and gradient profiles, are essential for reproducibility at scale.
- Formation and Reduction: Dissolve anthracycline 13-tosylhydrazone in anhydrous methanol with p-toluenesulfonic acid and reduce using sodium cyanoborohydride under reflux at 68-72°C.
- Purification and Protection: Isolate the 13-deoxy derivative via chromatography, then protect the amine functionality using di-tert-butyl dicarbonate to form the N-Boc derivative.
- Ammoniation and Deprotection: Treat the protected intermediate with ammonia at 0-4°C to form the imino group, followed by acidic removal of the Boc group to yield the final hydrochloride salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers substantial strategic benefits beyond mere technical novelty. The primary advantage lies in the simplification of the safety profile, which translates directly into reduced liability and broader market acceptance for the final drug product. By eliminating the cardiotoxic metabolic pathway, the resulting API allows for higher dosing regimens, potentially increasing the volume of active ingredient required per treatment course, thus driving demand for high-quality intermediates. Furthermore, the synthetic methodology relies on commodity chemicals such as sodium cyanoborohydride, p-toluenesulfonic acid, and standard organic solvents like methanol and chloroform. This reliance on readily available raw materials significantly enhances supply chain reliability, mitigating the risk of bottlenecks associated with scarce or highly regulated reagents. The process avoids the use of expensive transition metal catalysts, which not only lowers the direct material cost but also simplifies the downstream purification process by eliminating the need for rigorous heavy metal scavenging steps, a common cost driver in modern API manufacturing.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts and the use of standard acidic reduction conditions streamline the production workflow. By avoiding complex catalytic cycles and the associated ligand costs, the overall cost of goods sold (COGS) is significantly optimized. Additionally, the robust nature of the tosylhydrazone reduction allows for simpler reactor configurations compared to sensitive organometallic transformations, reducing capital expenditure requirements for specialized equipment. The purification strategy, while involving chromatography, utilizes standard silica and phenyl phases that are widely available and scalable, avoiding the need for custom-synthesized stationary phases that can inflate operational expenses.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, particularly the anthracycline precursors and tosylhydrazine derivatives, are established commodities within the fine chemical sector. This ensures a stable and continuous supply stream, reducing the lead time for high-purity pharmaceutical intermediates. The process does not depend on single-source suppliers for exotic reagents, providing procurement teams with the flexibility to qualify multiple vendors for raw materials. This diversification is critical for maintaining business continuity and protecting against market volatility or geopolitical disruptions that often impact the availability of specialized chemical reagents.
- Scalability and Environmental Compliance: The reaction conditions, specifically the reflux temperatures of 68-72°C, are well within the operational range of standard glass-lined or stainless steel reactors used in commercial-scale production. This thermal profile facilitates easy scale-up from pilot plant to multi-ton manufacturing without the need for cryogenic cooling or high-pressure vessels. Moreover, the solvent system, primarily consisting of methanol and chloroform, allows for efficient recovery and recycling through standard distillation processes. This capability supports environmental compliance initiatives by minimizing solvent waste discharge and reducing the overall carbon footprint of the manufacturing process, aligning with the sustainability goals of major pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these specialized anthracycline derivatives. Understanding these details is key for partners evaluating the feasibility of integrating this technology into their oncology pipeline. The answers are derived directly from the experimental data and theoretical framework provided in the patent documentation, ensuring accuracy and relevance for technical decision-makers.
Q: How does the 13-deoxy modification reduce cardiotoxicity compared to traditional doxorubicin?
A: Traditional anthracyclines like doxorubicin are metabolized into 13-dihydro metabolites which are strongly cardiotoxic. By removing the 13-keto group entirely (13-deoxy), this metabolic pathway is blocked, preventing the formation of the toxic metabolite and allowing for higher cumulative doses without heart failure risks.
Q: What are the critical reaction conditions for the cyanoborohydride reduction step?
A: The reduction must be performed under strictly acidic conditions (pH 7.0 or lower) using p-toluenesulfonic acid in anhydrous methanol. Temperatures should be maintained between 68-72°C; exceeding 72°C risks decomposition of both reactants and the sensitive anthracycline product.
Q: Why is the 5-imino group significant in this derivative class?
A: The 5-keto group in standard anthracyclines is associated with free radical generation, a secondary mechanism of cardiotoxicity. Converting this keto group to an imino function (5-imino) modifies the electronic structure to minimize free radical production, further enhancing the safety profile of the drug.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Imino-13-Deoxydoxorubicin Supplier
As the global demand for safer, more effective oncology treatments continues to rise, the ability to manufacture complex intermediates like 5-imino-13-deoxy anthracyclines with precision and consistency is paramount. NINGBO INNO PHARMCHEM stands at the forefront of this capability, leveraging extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the sensitive chemistry required for these transformations, ensuring that every batch meets stringent purity specifications and rigorous QC labs standards. We understand that the margin for error in oncology API synthesis is non-existent, and our commitment to quality assurance guarantees that your supply chain remains uninterrupted and compliant with international regulatory frameworks.
We invite you to collaborate with us to unlock the full potential of this patented technology for your drug development programs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and process constraints. By partnering with NINGBO INNO PHARMCHEM, you gain access to deep process knowledge and the agility to adapt synthesis routes for maximum efficiency. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how we can support your journey from clinical trials to commercial launch with reliable, high-quality intermediates.